10/10/2017
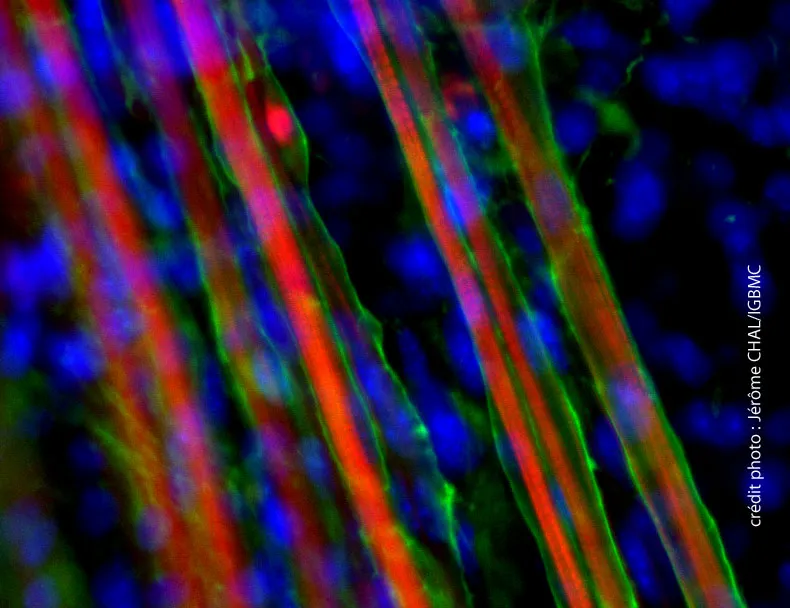
Maladie de Pompe à début tardif et enzymothérapie substitutive : l’expérience française
L’enzymothérapie de substitution est bien tolérée et est utile aux personnes adultes les plus sévèrement atteintes de glycogénose de type II à début tardif.