Glycogénoses musculaires
Tour d’horizon des différentes glycogénoses musculaires, de leur cause à leurs manifestations en passant par les traitements déjà disponibles et ceux, nombreux, qui sont en cours de développement pour ces maladies génétiques ayant pour point commun une accumulation de glycogène dans les cellules musculaires.

Une glycogénose musculaire, c’est quoi ?
Il existe plusieurs formes de glycogénoses. Ce sont toutes des maladies d’origine génétique qui se traduisent par une accumulation anormale d’une forme de réserve d’énergie dans les cellules : le glycogène. Cette accumulation peut concerner différents organes mais les plus souvent atteints sont ceux qui possèdent naturellement des réserves importantes de glycogène : les muscles (dont le muscle cardiaque) et le foie. Dans les glycogénoses musculaires, les symptômes sont liés à l’accumulation de glycogène dans les muscles, et aussi parfois dans le foie.
Un stock d’énergie inutilisable
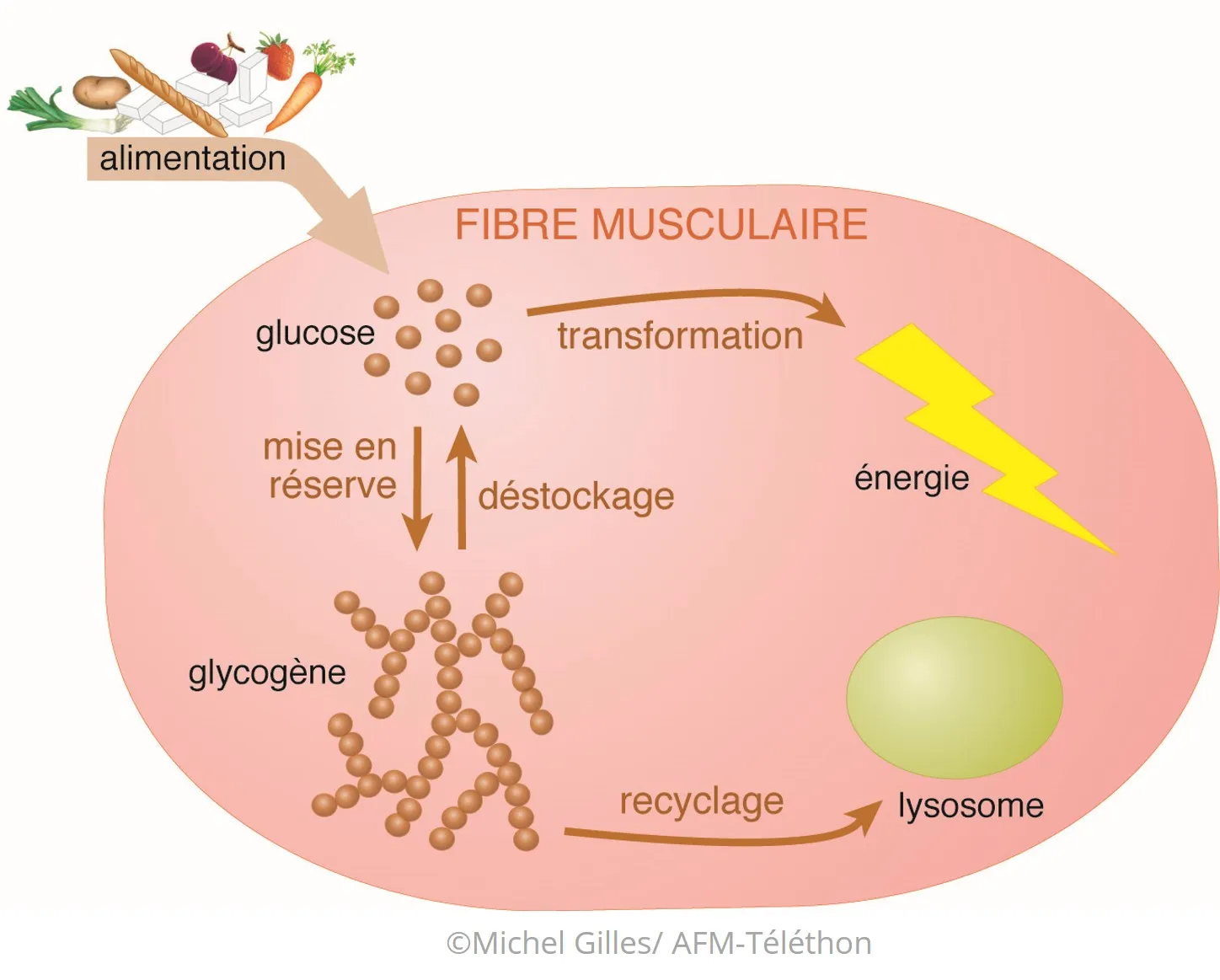
- Habituellement, les sucres (glucides) que nous mangeons et que nous n'utilisons pas tout de suite sont stockés dans les cellules du foie et des muscles sous forme de glycogène. Une seule molécule de glycogène se compose de milliers de molécules de glucose... jusqu’à 55 000 !
- Lors d’un exercice physique, du glycogène est transformé (déstockage) en énergie utilisable par les muscles grâce à une série de réactions biochimiques qui font intervenir plusieurs enzymes. Une partie du glycogène est aussi régulièrement recyclée, grâce à d'autres réactions.
- Si l’une de ces réactions ne se fait pas ou se fait mal, le glycogène s'accumule dans les cellules sans pouvoir être utilisé ou recyclé. C'est ce qui se passe dans les glycogénoses, où son accumulation peut perturber le fonctionnement de différents organes : muscles, foie, cœur, nerfs, cerveau...
Des symptômes musculaires variables
Les glycogénoses musculaires peuvent se manifester de façon très différente les unes des autres. Elles entrainent soit des symptômes lors des exercices physiques, comme une intolérance à l'effort, des douleurs musculaires (myalgies) ou des crampes, soit une faiblesse musculaire permanente.
Des maladies rares, voire ultra-rares
En France, la Banque nationale de données maladies rares (BNDMR) recense, pathologie par pathologie, les personnes suivies en centre expert. Selon ses chiffres publiés en Août 2025, les glycogénoses musculaires les plus fréquentes sont les Maladie de Pompe et de McArdle, qui rassemble chacune plus de 350 personnes consultant en centre spécialisé, suivies par les maladies de Cori-Forbes (autour de 200 personnes), puis de Danon et d’Andersen (une trentaine chacune).
La grande rareté des glycogénoses musculaires retarde souvent le diagnostic et donc la prise en charge, en particulier lorsque la maladie commence à se manifester à l’âge adulte.
Les formes néonatales, c’est-à-dire qui se manifestent dès les premiers jours de vie, se caractérisent le plus souvent par une baisse du tonus musculaire (hypotonie) généralisée. Sans traitement précoce, l'évolution est rapide et fatale.
Les formes plus tardives évoluent lentement. Certaines personnes développent une faiblesse musculaire permanente, notamment dans les muscles des épaules, des hanches, des bras et des jambes.
Quelle est la cause ?
Toutes les glycogénoses musculaires sont d’origine génétique : elles sont dues à des anomalies (mutations) de gènes qui codent des enzymes impliquées dans l’utilisation ou le recyclage du glycogène.
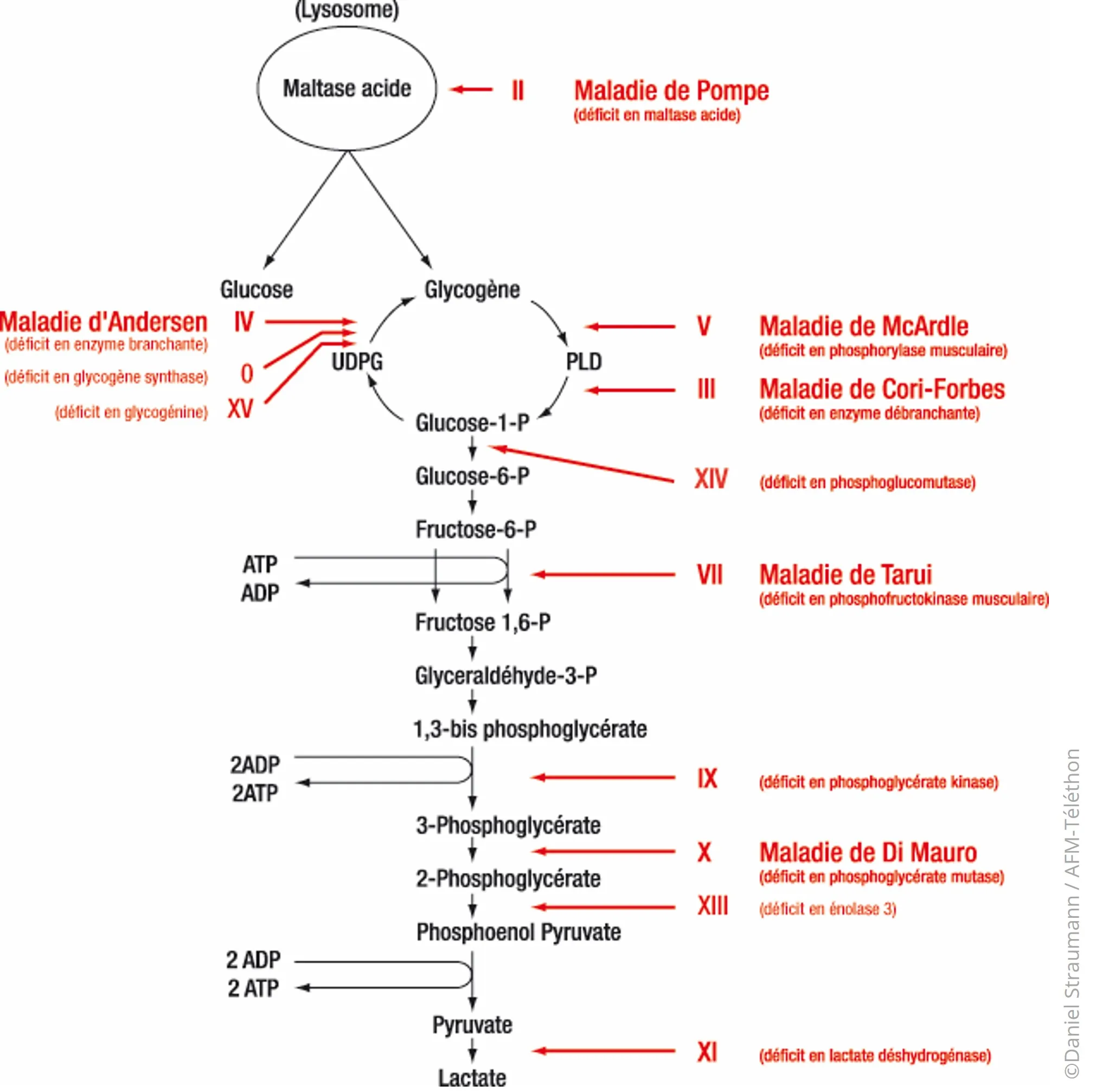
Les différentes formes de glycogénoses musculaires sont :
- type 0 ou déficit en glycogène synthase musculaire
- type II, maladie de Pompe ou déficit en alpha-glucosidase acide (ex-maltase acide)
- type III, maladie de Cori ou maladie de Forbes ou déficit en enzyme débranchante du glycogène
- type IV, maladie d'Andersen ou déficit en enzyme branchante du glycogène
- type V, maladie de McArdle ou déficit en phosphorylase musculaire
- type VII, maladie de Tarui ou déficit en phosphofructokinase
- type IX, ou déficit en phosphorylase kinase
- type X, maladie de Di Mauro ou déficit en phosphoglycérate mutase
- type XIII, ou déficit en bêta-énolase musculaire
- type XIV, ou déficit en phosphoglucomutase
- type XV, ou déficit en glycogénine-1
- maladie de Danon, due à un déficit en LAMP-2 (protéine associée à la membrane lysosomale 2)
Et les maladies à polyglucosans alors ?
Un polyglucosan est une forme de glycogène, mais de structure anormale. Il s’accumule sous forme de dépôts cellulaires dans différentes glycogénoses :
- les myopathies à polyglucosans, pour lesquels deux gènes ont déjà été mis en cause, GYG1 qui code la glycogènine 1 et GBE qui code l’enzyme branchante du glycogène. La plus fréquente de ces myopathies est la maladie d’Andersen, ou glycogénose de type IV.
- la maladie à inclusions de polyglucosans de l’adulte (ou APBD pour Adult polyglucosan body disease) est dûe à une mutation du gène GBE1, avec déficit en enzyme branchante du glycogène. Elle se manifeste par des troubles neurologiques : engourdissements des extrémités, spasticité, envie fréquente d’uriner...
Quel est le traitement ?
Le diagnostic permet de mettre en route une prise en charge adaptée, laquelle repose sur des mesures différentes selon le type de glycogénose musculaire : apporter l’enzyme manquante (enzymothérapie substitutive) dans la maladie de Pompe, limiter les efforts intenses, aménager des temps de repos, adapter son régime alimentaire, éviter les épisodes d’hypoglycémie...
Dans tous les cas, le conseil génétique permet de s’informer sur le risque de développer soi-même la maladie ou de transmettre le gène muté à ses futurs enfants.
Le suivi idéal s’appuie sur des consultations régulières dans un centre expert de proximité :
- spécialisé dans les maladies rares neuromusculaires (filière Filnemus)
- ou dans maladies héréditaires du métabolisme (filière G2M)
Des ressources utiles
•Pour la prise en charge et le suivi au long cours
Les protocoles nationaux de diagnostic et de soins (PNDS) sont des recommandations de bonnes pratiques destinés aux professionnels de santé. À ce jour, trois glycogénoses musculaires ont un PNDS :
- la maladie de Pompe
- la glycogénose de type III
- la maladie de McArdle
Chacun de ces documents débute par une « synthèse au médecin traitant » qui présente de façon simple et claire la maladie et le parcours de soins. N’hésitez pas à montrer cette synthèse à vos soignants !
•Pour les situations d’urgence
Le site Orphanet rassemble des recommandations spécifiques pour la prise en charge des personnes atteintes de différentes glycogénoses musculaires. Elles sont prioritaires et ne devraient pas attendre aux urgences lorsque la maladie entraine un risque d’hypoglycémie, par exemple suite à des vomissements ou un jeûne prolongé.
Où en est la recherche dans les glycogénoses musculaires ?
En chiffres
- 53 essais et études cliniques en cours ou en préparation dans différents pays pour les glycogénoses de tous types, répertoriés le 1er décembre 2025 sur ClinicalTrials.gov.
- 316 nouveaux articles médico-scientifiques sur les glycogénoses recensés au cours des 12 derniers mois par Pubmed (au 1er décembre 2025).

De nouveaux traitements à l’étude
Les chercheurs explorent différentes pistes thérapeutiques.
Apporter un gène fonctionnel pour suppléer le gène muté
Plusieurs produits de thérapie génique sont développés dans des glycogénoses musculaires.
- L’ACTUS-101 est actuellement à l’essai aux États-Unis dans la forme tardive de la maladie de Pompe, de même que l’AT845, évalué outre-Atlantique et au Royaume-Uni dans la même indication. Des résultats préliminaires présentés en mars 2025 aux États-Unis montrent des signes de transfert effectif au muscle du gène médicament (transduction) chez les quatre participants de l’essai AT845. Trois ont pu arrêter l’enzymothérapie de substitution 10, 17 et 24 semaines après cette thérapie génique et ne l’avaient pas repris deux ans après ce traitement.
- Dans la glycogénose de type III, Généthon travaille sur plusieurs approches pour apporter le gène GDE, qui est trop grand pour pouvoir être transporté dans un seul virus AAV :
- générer un mini-GDE, plus petit mais conduisant à la production d’une enzyme efficace chez des souris ou des rats atteints de la maladie ainsi que sur des cellules humaines issus de patients.
- utiliser deux vecteurs AAV pour transporter GDE, ce qui corrige l’accumulation de glycogène dans le foie et les muscles d’un modèle murin de la maladie, mais peut entrainer des réactions immunitaires. La combinaison de cette thérapie génique double avec la rapamycine, un immunosuppresseur, réduirait la réponse immunitaire.
- Aux États-Unis, sept personnes atteintes de la maladie de Danon ont participé à un essai de thérapie génique, laquelle a entrainé une amélioration : tous étaient dans un état stable au terme d’un suivi qui a duré jusqu’à 4 ans et demi.
Réduire l’accumulation de glycogène
Différentes équipes de recherche tentent de développer des produits qui empêchent la synthèse ou favorisent la dégradation du glycogène.
Il peut s’agir de petites molécules comme le guaiacol (ou gaïacol), le 144DG11 ou encore le MZ-101, qui ont fait l’objet d’études précliniques dans différentes glycogénoses, musculaires ou non, comme les maladies d’Andersen, de Pompe et à inclusions de polyglucosans de l'adulte. Le MZ-101 a également été évalué en association avec l’enzymothérapie de substitution, dans un modèle animal (souris) de maladie de Pompe. Le MZ-001, une petite molécule administrée sous forme de comprimés, cible également la maladie de Pompe. Un essai clinique conduit chez des volontaires non malades a montré que ce candidat-médicament est suffisamment bien toléré pour poursuivre son développement clinique.
Autre moyen de réduire les dépôts de glycogène, un petit ARN interférent (l’ABX1100) fait l’objet d’un essai de phase I qui inclut, depuis octobre 2024, des personnes atteintes de la forme tardive de la maladie de Pompe.
Mieux pallier le manque d’enzyme naturelle
Une enzymothérapie de substitution existe depuis plus de 20 ans dans la maladie de Pompe. Elle consiste à apporter, par perfusion, une enzyme fabriquée en laboratoire par génie génétique. Des études en cours s’attachent à améliorer son efficacité.
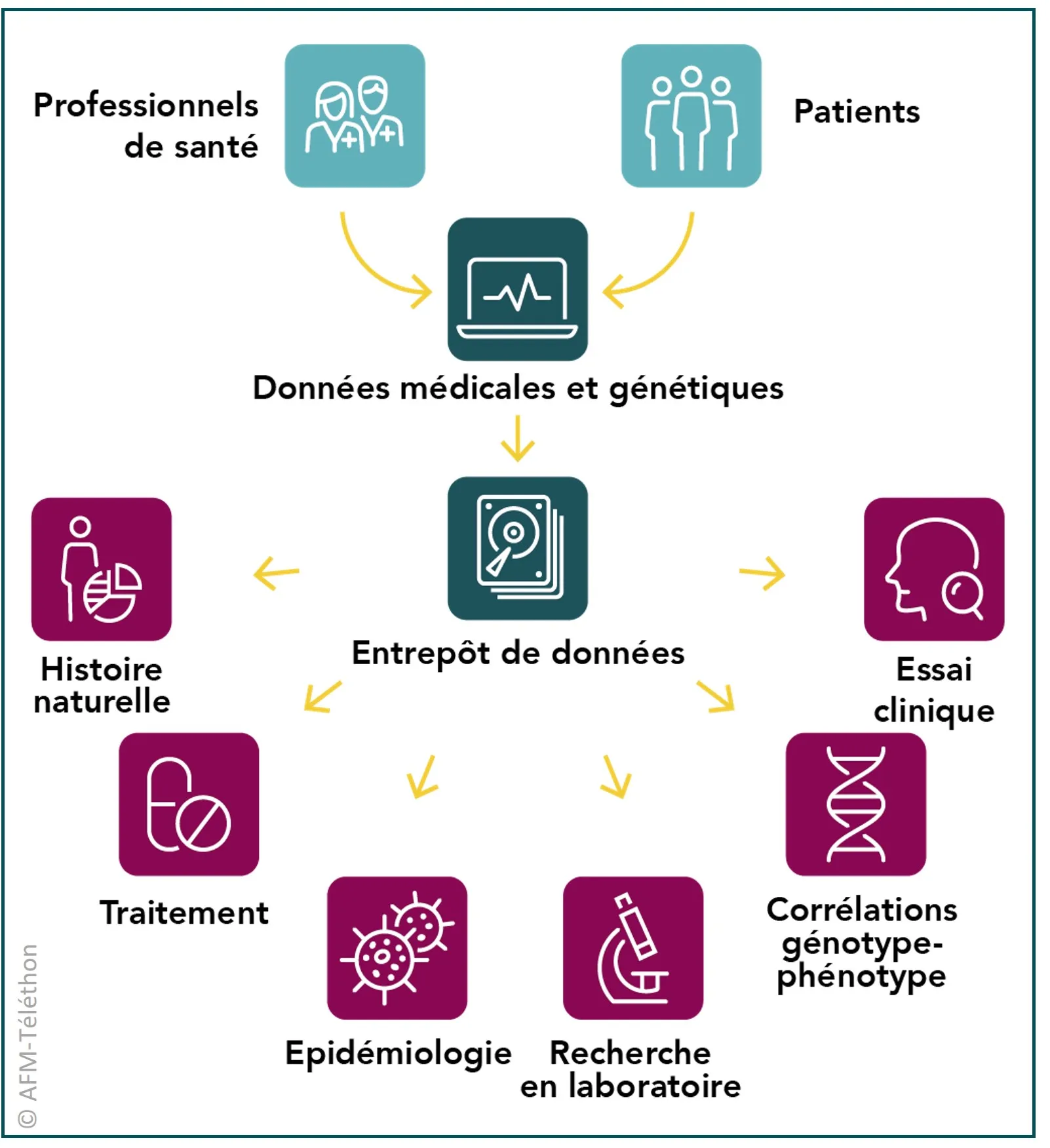
Des entrepôts de données
Chercheurs et médecins mettent en place des registres afin de recueillir des données de santé de personnes atteintes de glycogénoses musculaires. L’objectif est de faire progresser les connaissances, d’améliorer le diagnostic et les traitements. Il est également de faciliter le recrutement pour les essais cliniques.
- Le registre français de la maladie de Pompe (glycogénose de type II) a été créé en 2004, sous la responsabilité scientifique du Pr Pascal Laforêt (Hôpital Raymond Poincaré, Garches) et avec le soutien de l’AFM-Téléthon. En novembre 2024, ce registre recensait les données de 337 personnes.
- Une base de données Glycogénose de type III (maladie de Forbes ou de Cori) est née en 2014 d'une collaboration entre un Centre de référence Maladies neuromusculaires (Institut de Myologie, Paris) et un Centre de référence Maladies héréditaires du métabolisme hépatique (Hôpital Béclère, Clamart). L'objectif est d'inclure 150 participants.
- Un registre international Glycogénose de type V (maladie de McArdle) et autres glycogénoses musculaires avec intolérance à l'effort (hors maladie de Pompe) a été créé en 2013. Nommé EUROMAC registry, ce projet regroupe des données provenant de dix pays européens dont la France, et des États-Unis. Mi-2023, cette base de données recensait 282 patients dont 269 atteints de maladie de McArdle.


