19/08/2011
Dysferlinopathies et transplantation de mésangioblastes
L’expression de dysferline est partiellement restaurée par transplantation de mésangioblastes de souris dans un modèle murin de dysferlinopathie.
Numéro vert - Service et appel gratuits quel que soit votre opérateur
Numéro vert - Service et appel gratuits quel que soit votre opérateur
Retrouvez ici toutes les actualités concernant l'AFM-Téléthon et les maladies rares.
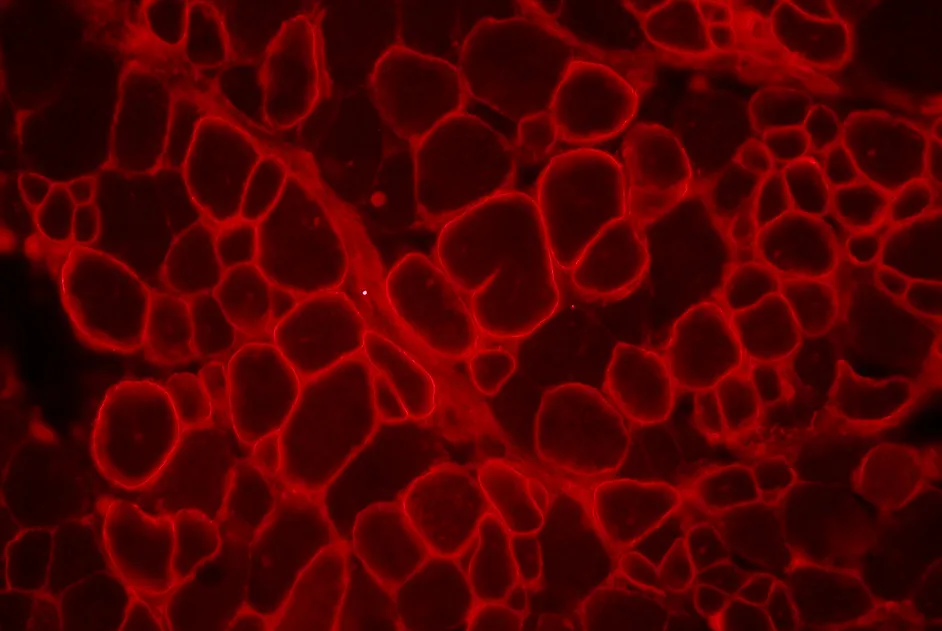
L’expression de dysferline est partiellement restaurée par transplantation de mésangioblastes de souris dans un modèle murin de dysferlinopathie.
Distraire un enfant qui subit un soin ou examen, est efficace pour réduire l’anxiété et la douleur.

Essais cliniques de médicaments autorisés dans l’Union européenne grâce à l’ouverture du Registre des essais cliniques.

Un registre créé par TREAT-NMD recueille les données de personnes ayant une mutation dans le gène FKRP dans le cadre d’études cliniques.

Description d’une nouvelle famille atteinte de myopathie congénitale à bâtonnets de type 6 et identification du gène en cause (gène KBTBD13).

Un comité international publie un consensus sur le diagnostic et la prise en charge des dystrophies musculaires congénitales.

Un comité international publie un consensus sur le diagnostic et la prise en charge des dystrophies musculaires congénitales.

Identification d’anomalies génétiques dans le gène TRAPPC11 à l’origine d'une myopathie des ceintures et d'une myopathie avec mouvements anormaux et déficience intellectuelle.

Preuve de concept de l’efficacité du saut d’un pseudo-exon du gène RYR1 pour augmenter l’expression de la protéine RYR1 normale.
Le saut d’un pseudo-exon dans le gène de la calpaïne 3 permet de corriger les conséquences de la présence anormale de ce pseudo-exon.
Publication du compte rendu du 184e atelier ENMC consacré à la douleur et à la fatigue dans les maladies neuromusculaires.
Le compte rendu du 198e workshop ENMC, correspondant au 7e atelier exclusivement consacré aux myopathies centronucléaires, est paru en août 2013.
Identification d’un nouveau gène à l’origine d'une myopathie congénitale, le gène HACD1.

Une thérapie génique avec de nouveaux AAV-calpaïne 3 améliore les symptômes de souris modèles de LGMD2A sans être toxique pour leur cœur.
Deux études publiées en juin 2013 rapportent l’identification de 2 nouveaux gènes à l’origine d’alpha-dystroglycanopathies : le gène GMPPB et le gène DPM1.