05/03/2014
SMA et ISIS-SMNRx : une annonce préliminaire
Deux communiqués de presse font état de premiers résultats des essais de l’ISIS-SMNRx en cours dans l’amyotrophie spinale proximale liée à SMN1.
Numéro vert - Service et appel gratuits quel que soit votre opérateur
Numéro vert - Service et appel gratuits quel que soit votre opérateur
Retrouvez ici toutes les actualités concernant l'AFM-Téléthon et les maladies rares.

Deux communiqués de presse font état de premiers résultats des essais de l’ISIS-SMNRx en cours dans l’amyotrophie spinale proximale liée à SMN1.

L’injection de 3 vecteurs AVV transportant chacun un fragment du gène de la dystrophine permet une expression de la protéine entière chez la souris.
Une étude confirme dans des souris modèles que SMN est plus importante pendant l’enfance (pour la maturation de la jonction neuromusculaire) qu’à l’âge adulte.
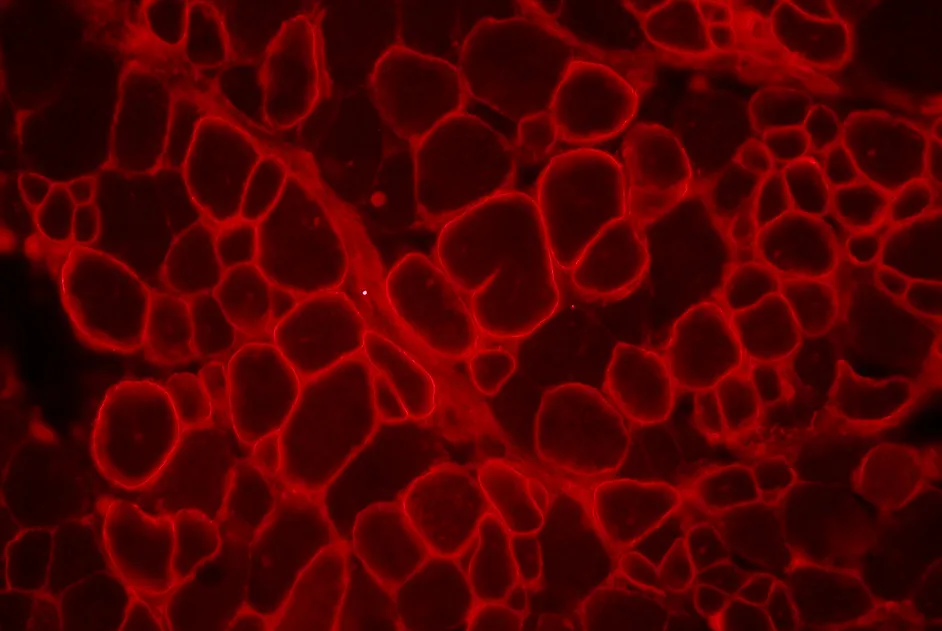
Des anticorps dirigés contre différentes parties de la dystrophine permettent de reconnaitre la dystrophine produites par des fibres révertantes.
Chez les patients qui n’ont pas une perte complète du gène SMN1, la sévérité de la maladie n’est pas corrélée au nombre de copies de SMN2.
L’administration d’oligonucléotides antisens associé à un peptide cible mieux le cœur d’une souris modèle de myopathie de Duchenne.

L’expression de la dystrophine est augmentée dans les muscles de 2/3 des participants à l’essai de phase IIa de l’ataluren.
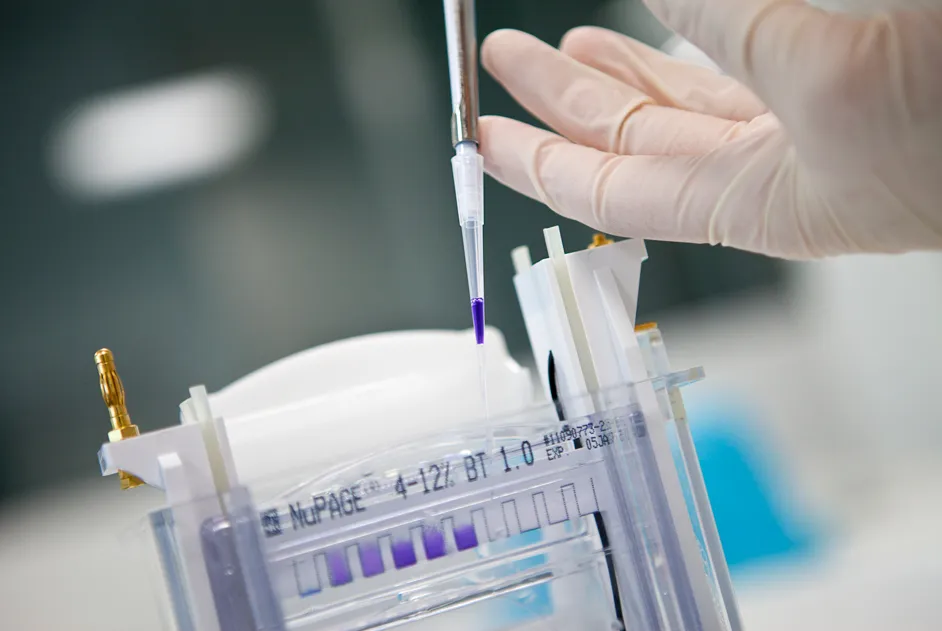
La tolérance et l’innocuité de trois doses de SMT C1100 sont évaluées au Royaume-Uni dans un essai de phase Ib chez 12 garçons atteints de DMD.
Absence de bénéfice de l'hormone de croissance sur la force musculaire de 19 patients atteints d’amyotrophie spinale proximale liée à SMN1 de type II ou III
Implication du gène du gène FBX038 à l’origine d’une amyotrophie spinale distale prédominant aux mollets.

Une nouvelle méthode de thérapie génique utilisant 3 AVV transportant chacun un fragment du gène de la dystrophine permet l’expression de la protéine entière.

L’expression de l’utrophine, une protéine proche de la dystrophine qui pourrait la remplacer dans la myopathie de Duchenne, est augmentée par FHL1.

Une étude d’histoire naturelle et de corrélation génotype/phénotype, soutenue par l’AFM-Téléthon, a été réalisée à partir de la banque UMD-DMD France.
Publication des résultats de l'essai de phase I du drisapersen chez 20 garçons atteints de myopathie de Duchenne non marchants

L’agence américaine FDA autorise le démarrage, aux États-Unis, d’un essai de thérapie génique de phase I dans l’amyotrophie spinale de type I.