Canalopathies musculaires
Les canalopathies musculaires sont dues à des anomalies génétiques dans un canal ionique perturbant la contraction du muscle. Le caractère fluctuant des manifestations complique et retarde l’identification de la maladie. Or, connaitre le diagnostic est essentiel pour bénéficier de conseils et de médicaments adaptés. Ce sont des maladies chroniques qui doivent bénéficier d’un suivi spécialisé.
Une canalopathie musculaire, c’est quoi ?
Les canalopathies musculaires sont des maladies très rares qui touchent aussi bien les enfants que les adultes.
Des origines génétiques
Elles sont toutes dues à une anomalie (mutation) d’un gène qui code un canal ionique, d’où leur nom de « canalopathie ». Cette anomalie se transmet selon un mode récessif ou dominant.
Le saviez-vous ?
Un canal ionique est une protéine insérée dans la membrane des cellules et qui la traverse. Il permet l’entrée et la sortie dans la cellule musculaire d’un ion donné : chlore (Cl-), sodium (Na+), calcium (Ca2+) ou potassium (K+). Les différents canaux ioniques jouent un rôle déterminant dans la contraction et le relâchement des muscles.
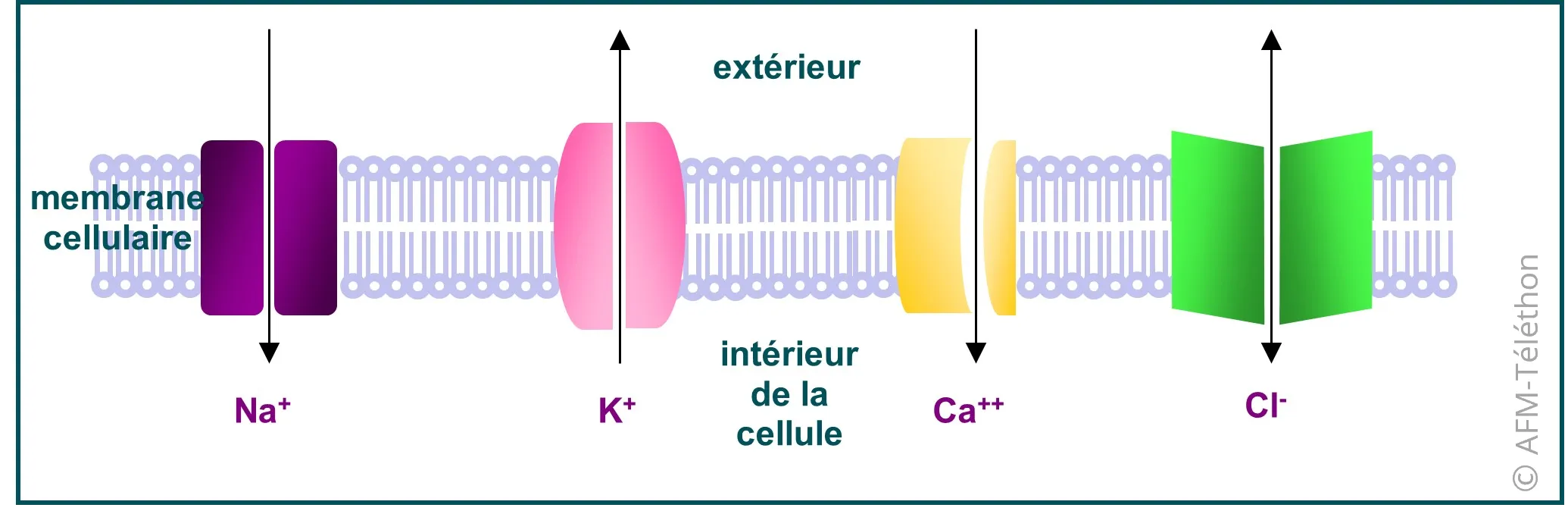
Une canalopathie musculaire peut impliquer un canal sodium (codé par le gène SCNA4), un canal potassium (codé par les gènes KCNJ2 ou KCNJ5), un canal calcium (codé par le gène CACN1AS) ou un canal chlore (codé par le gène CLNC-1). L’un de ces cinq gènes présente une anomalie génétique, conduisant au dysfonctionnement du canal ionique correspondant.
Deux grandes familles
Il existe deux grands types de canalopathies musculaires :
- les paralysies périodiques, pour lesquelles les muscles ont du mal à se contracter (hypoexcitabilité); elles concernent une centaine de familles en France.
- les syndromes myotoniques non dystrophiques, où les muscles ont du mal à se relâcher (hyperexcitabilité); ils touchent environ un millier de personnes en France.
Les paralysies périodiques comportent :
- les paralysies périodiques hypokaliémiques de type 1 et de type 2,
- la paralysie périodique hyperkaliémique,
- la paralysie périodique normokaliémique,
- le syndrome d’Andersen-Tawil.
La kaliémie correspond au taux de potassium (K+) dans le sang (« émie »). Certaines paralysies périodiques se manifestent par des accès de faiblesse musculaire associés à un taux de potassium anormalement élevé (hyperkaliémie) ou anormalement bas (hypokaliémie).
Les syndromes myotoniques non dystrophiques comportent :
- la myotonie congénitale de Thomsen
- la myotonie congénitale de Becker
- la paramyotonie congénitale (ou paramyotonie de von Eulenburg)
- les myotonies du canal sodium, dont font partie les myotonies aggravées par le potassium et sensible à l’acétazolamide.
Quels sont les symptômes ?
Les canalopathies musculaires sont de maladies chroniques, dont les premiers manifestations surviennent le plus souvent dans l’enfance ou à l’adolescence. Elles se manifestent dans la plupart des cas de façon intermittente, par :
- des crises de paralysie ou de faiblesse musculaire, qui durent de quelques minutes à plusieurs jours, pour une paralysie périodique ; le syndrome d’Andersen-Tawil peut se manifester aussi par des troubles du rythme cardiaque, qui nécessite un suivi par un cardiologue spécialisé,
- une raideur musculaire ou des difficultés à relâcher les muscles après un mouvement (myotonie), pour un syndrome myotonique non dystrophique.
L’intensité des symptômes est très variable d’une personne à l’autre. Une gêne respiratoire ou des difficultés à avaler sont possibles, si la paralysie ou la myotonie concerne les muscles respiratoires ou ceux du pharynx.
La paralysie ou la raideur s’accompagne parfois de douleurs musculaires et concerne un ou plusieurs muscles (membre, cou, thorax…). Certains facteurs peuvent favoriser la survenue de ces symptômes ou augmenter leur intensité. Selon la forme de canalopathie musculaire, ces facteurs peuvent être : l’exposition au froid, le jeûne, les repas riches en sucres (produits sucrés, féculents…), l’humidité, les aliments riches en potassium, la fièvre, le stress ou encore la grossesse.
Des manifestations souvent atypiques dans le syndrome d’Andersen-Tawil
En France, sur 35 personnes suivies pour un syndrome d’Andersen-Tawil en centre expert, seules 15 présentent des signes typiques tels que des accès de paralysie associés à des troubles du rythme cardiaque et à une morphologie particulière du visage et des doigts.
Lire l'actualité sur les symptômes atypiques dans ce syndrome.
Des maladies volontiers peu évolutives
- Dans les paralysies périodiques, l’intensité de la faiblesse musculaire varie d’une crise à l’autre chez une même personne. La fréquence et l’importance des crises de paralysie ont tendance à s’estomper avec l’âge pour parfois faire place à une faiblesse musculaire modérée permanente.
- Dans les syndromes myotoniques non dystrophiques, les symptômes évoluent peu au cours de la vie. Néanmoins, l’intensité de la raideur peut fluctuer d’un jour à l’autre, en fonction des circonstances (stress, émotions, froid, jeûne..). Il se développe parfois une hypertrophie des muscles concernés.
Quel est le traitement ?
Le diagnostic et la prise en charge d'une canalopathie musculaire se conçoivent dans le cadre de consultations pluridisciplinaires spécialisées dans les maladies neuromusculaires.
La prise en charge a pour objectif de réduire l’impact de la maladie sur la vie quotidienne et de prévenir les complications. Elle est déterminée au cas par cas, en fonction du type de canalopathie, de la gêne qu’elle entraine et de la fréquence des symptômes.
Un Centre de référence national
Spécialisé dans les canalopathies musculaires, ce centre est situé à l’Hôpital de la Pitié-Salpêtrière (Paris). Son équipe travaille en collaboration avec le réseau des consultations pluridisciplinaires et lui apporte, si nécessaire, son expertise à l’étape du diagnostic, du traitement et du suivi.
Le Centre national de référence des canalopathies musculaires exerce également des missions de recherche (coordination de RESOCANAUX, animation d’essais cliniques), de formation et d’information des professionnels de santé et des personnes malades. Il lui revient enfin de définir, faire évoluer et diffuser les recommandations de diagnostic et de prise en charge, au fur et à mesure des progrès de la recherche.
Voir le site internet du Centre de référence des canalopathies musculaires
Un groupe international d’experts, dont un médecin du Centre national de référence des canalopathies musculaires, a publié en mai 2023 des recommandations pour le diagnostic et la prise en charge des enfants atteints de canalopathies musculaires.
Des recommandations pour la surveillance cardiaque des adultes atteints de canalopathie sous mexilétine (Namuscla), le traitement de première intention de la myotonie, ont été émises par huit experts de différents Centres de références français ainsi qu’un pharmacologue italien.
Traiter les crises de paralysie périodique
Les mesures à prendre sont spécifiques à chaque paralysie périodique.
Pour une forme hyperkaliémique, l’intensité et/ou la durée de la crise peut être réduit par la prise d’un aliment ou d’une boisson sucrée et la pratique d'une activité physique légère (marche), dès l’apparition de la faiblesse musculaire, ou éventuellement par la prise d’un médicament (salbutamol).
En cas de paralysie périodique hypokaliémique, le traitement de la crise repose sur la prise de potassium et il faut éviter tout apport sucré concomitant (boisson ou aliment sucré, perfusions glucosées…).
Les paralysies périodiques sont des maladies très rares, c'est pourquoi elles sont mal connues par les médecins qui ne sont pas experts des maladies neuromusculaires. Vous pouvez demander au spécialiste qui vous suit de rédiger et de vous confier un document qui explique, à tout autre médecin que vous seriez amené à consulter, la conduite à tenir en cas d’accès paralytique.
Le traitement de fond
- Il a pour objectif de prévenir la survenue des symptômes ou d’en réduire l’intensité, lorsqu’ils sont fréquents et gênants. Ce traitement peut reposer sur différents médicaments : mexilétine, carbamazépine, acétazolamide, spironolactone, dichlorphénamide…
La mexilétine (Namuscla) bénéficie d'une autorisation de mise sur le marché (AMM) dans le traitement symptomatique de la myotonie chez les adultes atteints de troubles myotoniques non dystrophiques.
- En complément, des précautions simples sont à prendre au quotidien, pour éviter ou limiter les facteurs qui favorisent ou aggravent les symptômes. Selon la forme de canalopathie, il peut s’agir d’éviter le jeûne, les aliments salés, riches en sucres ou en potassium, les efforts en milieux froids, ou encore les efforts intenses.
- Des séances de kinésithérapie (massages, infrarouges, étirements, balnéothérapie) permettent de lutter contre les douleurs musculaires et les rétractions des tendons.
- Les personnes atteintes d’un syndrome d’Andersen-Tawil doivent bénéficier d’un suivi par un cardiologue spécialisé dans les maladies neuromusculaires afin de mettre en route un traitement adapté, voire de bénéficier d’un défibrillateur cardiaque implantable, si une anomalie est détectée. Il faut également éviter de prendre certains médicaments, dont la liste peut être téléchargée sur le site du Centre de référence national.
Une carte pour les situations d'urgence
Les cartes d’urgence "Syndromes myotoniques non dystrophiques" et "Paralysie périodique hypokaliémique", mises en place par la filière FILNEMUS , présentent les recommandations importantes pour la sécurité et la prise en charge médicale d’urgence du malade.
Des précautions anesthésiques
Une canalopathie n’empêche pas de bénéficier d’une anesthésie générale ou d'une péridurale, mais il faut prendre des précautions particulières. Certains produits anesthésiants et certaines circonstances propres à une intervention chirurgicale (froid, jeûne, perfusion de glucose…) peuvent, en effet, provoquer ou aggraver les symptômes. Quelle que soit l’intervention envisagée, il faut donc toujours prévenir l’anesthésiste et le chirurgien que l’on est atteint d’une canalopathie musculaire et leur présenter la Carte d’urgence. L’anesthésiste peut aussi contacter l’équipe du Centre national de référence des canalopathies musculaires ou consulter les recommandations publiées sur leur site internet.
Où en est la recherche?
La recherche sur les canalopathies musculaires est particulièrement active en France. Une équipe dirigée par Bertrand Fontaine, et soutenue par l'AFM-Téléthon, travaille spécifiquement sur ces maladies. Elle anime également un réseau spécialisé sur les canalopathies musculaires, RESOCANAUX, et travaille notamment en collaboration avec des experts du Canada, des États-Unis, de la Grande Bretagne, des Pays-Bas et de l'Italie.
L’objectif des chercheurs est à la fois de mieux comprendre ces maladies (fonctionnement des canaux ioniques, mutations génétiques, mécanismes pathologiques pour chaque canalopathie…), évaluer l'impact des symptômes sur la qualité de vie des patients et d’étudier des candidats-médicaments, sur des modèles biologiques (en laboratoire) puis chez l'homme (dans le cadre d'essais cliniques).
Des essais cliniques de la mexilétine
La mexilétine (Namuscla), un agent bloquant des canaux sodium, permet de diminuer la raideur et les difficultés de décontraction musculaires (myotonie). Bénéficiant d'une autorisation de mise sur le marché dans le traitement symptomatique de la myotonie chez les adultes atteints de troubles myotoniques non dystrophiques, elle a fait l'objet d'un essai clinique, en France, auprès d'enfants et d'adolescents. Celui-ci est terminé, les données sont en cours d'analyse.
Un autre essai clinique est en cours en France pour évaluer les effets de la mexilétine chez des adultes sur le long terme.
Les connaissances génétiques progressent
En règle générale, une paralysie périodique résulte d’une seule mutation du gène SCN4A, qui code le canal sodium. Des médecins japonais ont récemment décrit le cas d’une famille atteinte d’une forme atypique de paralysie périodique hypokaliémique, avec deux mutations (dont une nouvelle) de SCN4A.
Un ancien anti-épileptique efficace chez des souris
Aux États-Unis, des chercheurs ont montré que la rétigabine prévient de façon efficace les accès de faiblesse musculaire dans deux souris modèles de paralysie périodique hypokaliémique. Cette molécule était commercialisée jusqu’en 2017, sous le nom de Trobalt®, pour prévenir les crises d’épilepsie. Elle provoque l’ouverture des canaux potassium. Dans la paralysie périodique hypokaliémique, les accès de paralysie sont causés par un état temporaire d’inexcitabilité de la fibre musculaire. Le retour à la normale est favorisé par une augmentation de la perméabilité des fibres musculaires au potassium, ce que favoriserait la rétigabine.
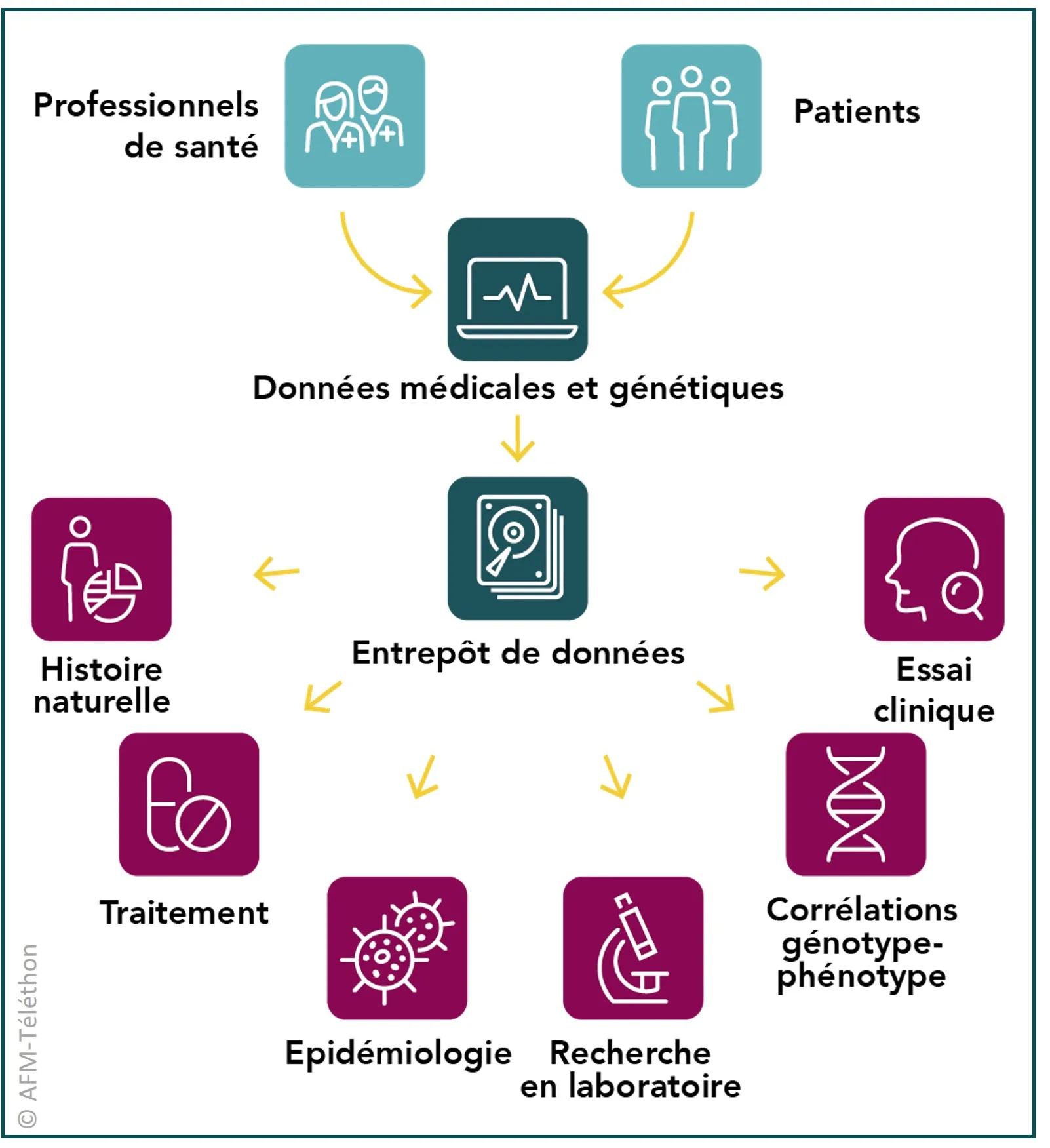
Une base de données française
En France, le réseau spécialisé dans les canalopathies musculaires RESOCANAUX a développé une base de données nationale pour recueillir les données génétiques et médicales des personnes atteintes de ces maladies, avec l’objectif de faire progresser les connaissances (nombre de patients, symptômes, évolution…) et de faciliter le recrutement à de futurs essais cliniques.



Comment faire le diagnostic ?
L’électromyogramme aide à confirmer le diagnostic de canalopathie musculaire, et à préciser sa forme. Cet examen comporte un enregistrement de l’activité électrique du muscle, au repos et lors de différents tests (efforts, froid).
Une prise de sang permet de réaliser un test génétique, à partir de l’ADN des globules blancs, pour rechercher l’anomalie génétique en cause. La canalopathie musculaire peut rester « non déterminée » à l’issue de ce test, car toutes les anomalies génétiques responsables de canalopathie n’ont pas encore été découvertes.
Pensez-y
Le conseil génétique s'adresse aux personnes concernées par une maladie d'origine génétique, à l’exemple d’une canalopathie musculaire, qu'elles soient elles-mêmes atteintes ou que l’un de leur proche le soit. L’objectif est de les informer sur le risque qu'elles ont de développer et/ou de transmettre cette maladie.