Dystrophies musculaires congénitales
Les dystrophies musculaires congénitales sont des maladies génétiques rares qui affaiblissent les muscles très tôt dans l’enfance, voire dès la naissance. Des soins précoces et soutenus aident à limiter les conséquences de la maladie sur la motricité, la respiration, le fonctionnement cardiaque… pour préserver le développement harmonieux de l’organisme.
Différentes formes de DMC
Les dystrophies musculaires congénitales (DMC) sont caractérisées par une faiblesse musculaire apparente, dans la majorité des cas, dès la naissance (congénitale) ou dans les six premiers mois de la vie, et toujours avant l’acquisition de la marche.
Ces maladies génétiques forment un groupe hétérogène : plus d’une trentaine de gènes différents en cause dans les DMC ont été identifiés. Ces gènes jouent des rôles dans le fonctionnement des cellules musculaires, la structure de leur environnement proche (la matrice extracellulaire), ou encore l’ancrage des fibres musculaires dans leur milieu.
Actuellement, on distingue six groupes de DMC :
- Collagénopathies : aussi appelées DMC de type Ullrich, elles sont liées à des anomalies des gènes du collagène (COL6A1, A2, A3 et COL12A1).
- Mérosinopathies primaires : dues à des anomalies du gène LAMA2 (DMC avec déficit en mérosine, aussi appelées DMC liée à LAMA2 ou DMC 1A).
- Alpha-dystroglycanopathies : aussi appelées dystroglycanopathies, elles sont liées à des anomalies de l’alpha-dystroglycane, protéine de la surface des cellules musculaires. Parmi les nombreux gènes en cause, la majorité joue un rôle dans la glycosylation de l’alpha-dystroglycane, processus nécessaire à son bon fonctionnement. Le groupe inclut entre autres le syndrome de Walker-Warburg (WWS), le syndrome muscle-œil-cerveau (syndrome MEB) et la DMC de type Fukuyama (DMCF).
- Laminopathies : liées à des anomalies du gène LMNA, qui code les lamines A/C.
- Sélénopathies : aussi appelées « syndrome de la colonne raide », elles sont dues au déficit en sélénoprotéine N lié à des anomalies du gène SELENON (anciennement SEPN1).
- Les autres DMC : groupe hétérogène dont les formes sont ultra-rares (moins d’une personne touchée sur 50 000). On y retrouve les DMC liées aux gènes BET1, GOSR2, TRAPPC11, INPP5K, CHKB, ITGA7, RYR1, SYNE1… impliqués dans le transport des vésicules cellulaires, le fonctionnement des mitochondries, etc.
À quoi sont-elles dues ?
Une grande partie des protéines codées par les gènes impliqués dans les DMC intervient dans la cohésion des fibres musculaires entre elles et avec leur environnement immédiat (matrice extracellulaire).
La laminine alpha 2 (mérosine), le collagène VI, le collagène XII, les intégrines et l’alpha-dystroglycane participent, chacune à leur niveau, au maintien d’une liaison entre l’intérieur et l’extérieur de la cellule. Ce système d’amarrage entre les deux milieux permet aux cellules musculaires de s’adapter aux contraintes mécaniques, en particulier aux déformations qu’elles subissent lors d’une contraction du muscle. Si ce lien est altéré, le muscle est alors fragilisé.
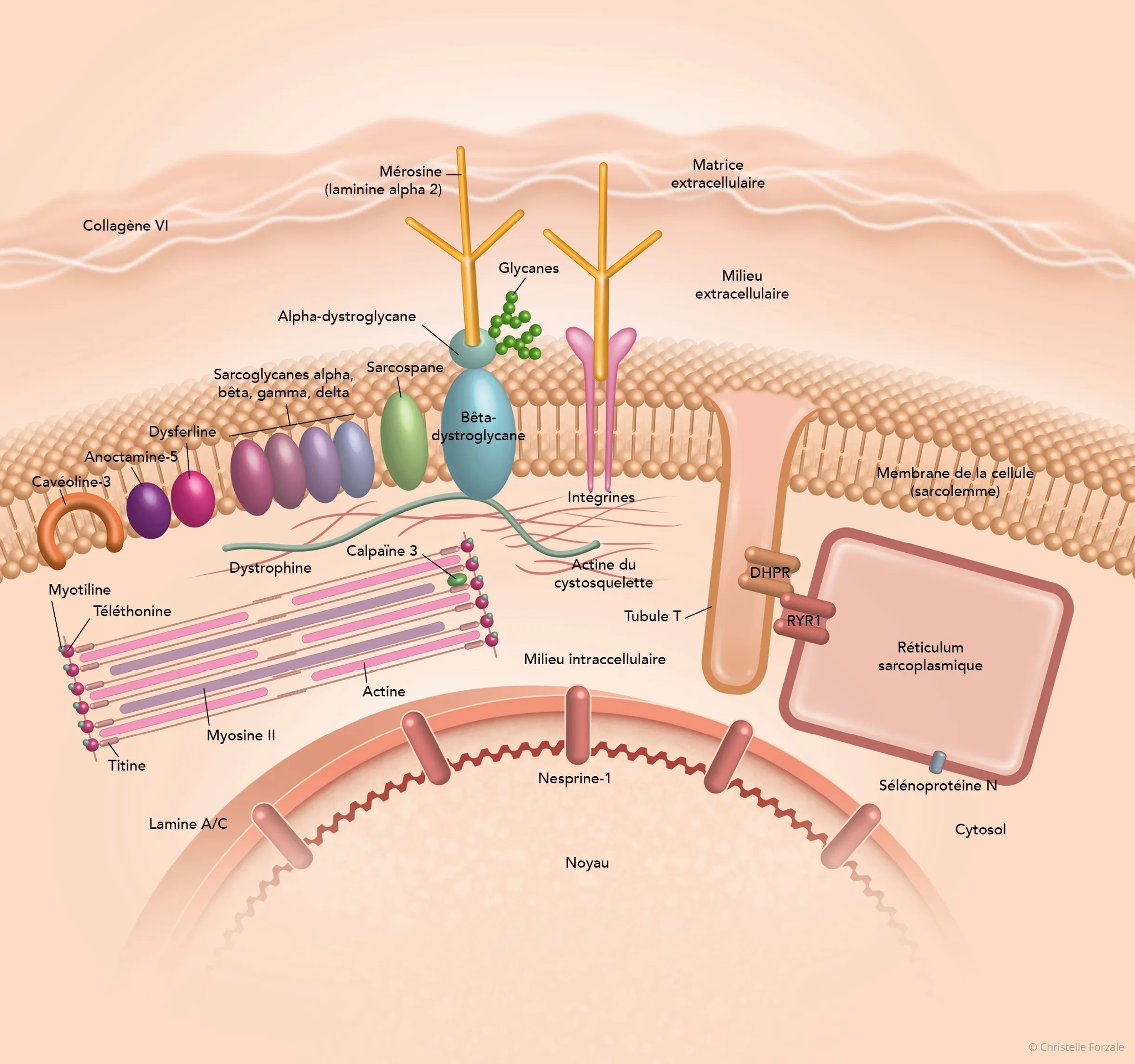
Gros plan sur des protéines impliquées dans les DMC.
Des maladies rares, voire « ultra-rares »
La fréquence globale des DMC est estimée entre 1 et 9 personnes pour 100 000. En France, on estime qu’il y aurait moins de 1000 malades atteints de DMC. Les collagénopathies, les mérosinopathies et les alpha-dystroglycanopathies sont les plus fréquentes, et constituent chacune un peu moins d’un tiers des malades. Les autres (laminopathies, sélénopathies…) sont beaucoup plus rares et ne concernent que quelques dizaines de malades dans le pays.
Quelles sont les manifestations ?
La maladie se manifeste par un manque de force et de tonus musculaires (hypotonie) dès la naissance. L’enfant a des difficultés d’acquisition de fonctions motrices comme se tenir assis, garder la tête droite, rester debout ou marcher. Plus tard, il peut avoir des difficultés à courir, chuter fréquemment…
Les muscles et les tendons ont tendance à s’enraidir, parfois avant la naissance, ce qui provoque fréquemment des déformations des membres (les médecins parlent d’arthrogrypose) et de la colonne vertébrale (dos creux, scoliose…).
Une insuffisance respiratoire peut apparaître de façon transitoire à la naissance, ou plus durablement chez l’adolescent ou le jeune adulte. Elle est due à l’atteinte des muscles respiratoires qui retentit aussi sur la croissance de la cage thoracique et le développement des poumons.
Une atteinte cardiaque est susceptible de survenir dans tous les types de DMC. Elle est cependant plus fréquente dans les dystroglycanopathies néonatales et dans les laminopathies, et plus rare et plus tardive dans les mérosinopathies et les sélénopathies.
Les formes les plus sévères de DMC peuvent s’accompagner d’anomalies oculaires, d’épilepsie, ou encore de difficultés intellectuelles.
La gravité de la maladie est variable d’une forme de DMC à l’autre, voire d’une personne à l’autre, même dans une même famille. La plupart sont assez peu évolutives et/ou à un rythme lent. Les progrès réalisés en matière de diagnostic et de prise en charge ont permis d’améliorer la qualité et l’espérance de vie des malades.
Quel suivi médical ?
Le diagnostic et la prise en charge d’une dystrophie musculaire congénitale se font au mieux dans des consultations pluridisciplinaires spécialisées dans les maladies neuromusculaires.
Les traitements sont pour le moment symptomatiques : la prise en charge vise à prévenir les complications, en particulier au niveau des muscles, des articulations, de l’appareil respiratoire, du cœur et du tube digestif, et à améliorer le confort de vie des malades. Elle est adaptée à la sévérité des symptômes.
Des recommandations de soins internationales
Un comité international de plus de 80 experts, l’International Standard of Care Committee for Congenital Muscular Dystrophy, s’est constitué, grâce au soutien financier de TREAT-NMD, Cure CMD et de l’AFM-Téléthon pour élaborer des recommandations de diagnostic et de prise en charge des dystrophies musculaires congénitales. Ces recommandations, qui font référence, ont été publiées en novembre 2010 (une version traduite en français est disponible).
- La prise en charge orthopédique entretient la souplesse et l’amplitude des mouvements ; elle prévient ou limite la progression d’une déformation de la colonne vertébrale (scoliose) par le port d’un corset et/ou d’une intervention chirurgicale (arthrodèse).
- La prise en charge respiratoire est commencée dès que le diagnostic est posé. Elle permet de faciliter le développement des poumons pour optimiser les capacités respiratoires puis d’entretenir une fonction pulmonaire optimale. La lutte contre l’insuffisance respiratoire chronique permet de limiter l’apparition d’une atteinte cardiaque secondaire.
- Un suivi cardiologique, précoce et régulier, permet de proposer des traitements adaptés (inhibiteurs de l’enzyme de conversion, diurétiques, défibrillateur…) dès les premiers signes d’une éventuelle atteinte cardiaque.
Des cardiologues spécialistes des DMC ont élaboré, avec des représentants AFM-Téléthon de personnes atteintes de DMC, un document de référence sur la prise en charge cardiologique de ces maladies, destiné aux professionnels de santé, pour les guider dans les soins cardiologiques de ces maladies.
- La prise en charge digestive et nutritionnelle améliore le transit, les éventuelles difficultés pour mastiquer ou avaler et prévient la dénutrition.
- En cas de faiblesse des muscles du visage, une prise en charge orthodontique favorise souplesse et croissance harmonieuse de la mâchoire et de la face.
- En cas d’atteinte cérébrale, une prise en charge spécifique (psychomotricité, orthophonie...) favorise le développement de l’enfant.
Les DMC étant des maladies génétiques, le conseil génétique permet de s’informer et d’être accompagné sur les risques de développer ou de transmettre la maladie.
Une recherche active
La recherche sur les DMC est internationale, avec une implication particulièrement forte en France, en Angleterre, en Allemagne, en Turquie, au Japon et aux États-Unis.
La France a été en pointe très tôt dans ce domaine, avec la découverte de la mérosine par l’équipe de Fernando Tomé et Michel Fardeau en 1994.
Quelques chiffres…
- Plus de 30 gènes impliqués identifiés. Les progrès dans les techniques d’analyse d’ADN permettent de continuer à découvrir de nouveaux gènes plus rarement en cause.
- 11 études cliniques en cours dans les dystrophies musculaires congénitales répertoriés sur le site ClinicalTrials.gov (interrogation du 01/12/2025).

Des pistes thérapeutiques à l’étude
Des étapes clés pour produire une protéine
C’est le gène, une séquence spécifique d’ADN dans le noyau de la cellule, qui contient l’information nécessaire pour réaliser une protéine. Cette information est d’abord retranscrite au sein d’une autre molécule, l’ARN.
- Celle-ci est transformée pour devenir un ARN messager, ou ARNm.
- L’ARNm est transporté hors du noyau et les instructions de fabrication qu’il contient sont traduites par la cellule pour faire la protéine.

Plusieurs stratégies thérapeutiques sont à l’étude afin d’agir sur les différents mécanismes pathologiques propres à chaque forme de DMC.
- l’ADN : avec l’introduction d’une copie fonctionnelle du gène muté responsable de la maladie, comme dans la DMC liée à FKRP, ou l'activation d’un « gène de secours », comme avec la « surexpression » de la laminine 111 dans la DMC liée à LAMA2.
- l’ARN messager : par exemple pour diminuer les quantités d’un gène muté devenu toxique, comme dans les DMC liées au collagène VI.
- La protéine : comme avec la restauration de la glycosylation de l’alpha-dystroglycane dans les alpha-dystroglycanopathies.
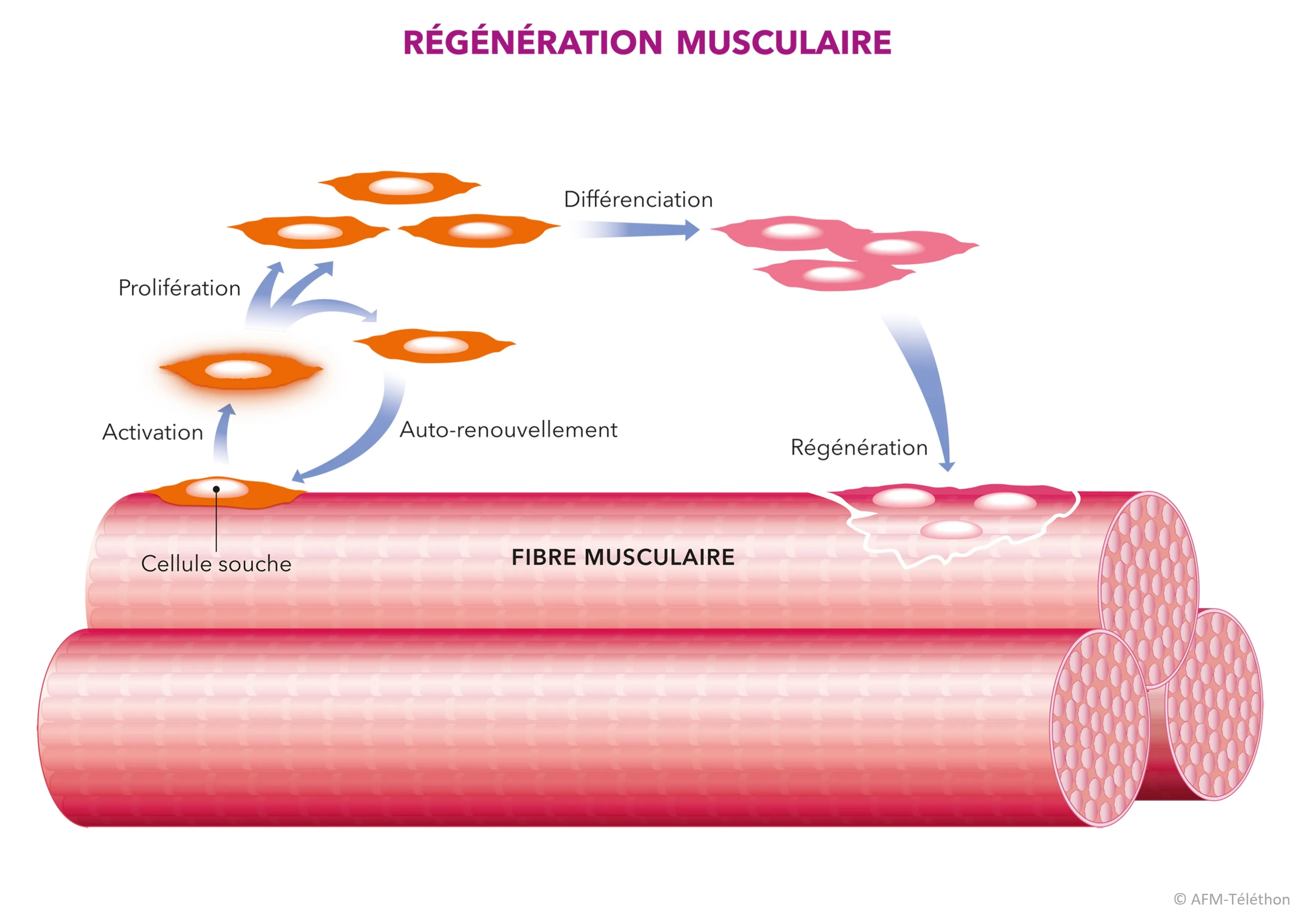
D’autres pistes thérapeutiques sont étudiées, comme le rétablissement de la connexion déficiente entre la cellule musculaire et son environnement proche, notamment dans les DMC liées à LAMA2, ou encore l’altération des cellules satellites pour aider le muscle à se régénérer.
VOIR LES AVANCÉES 2025 DANS LES DYSTROPHIES MUSCULAIRES CONGÉNITALES
Mieux connaître pour mieux soigner
- Les études d’histoire naturelle suivent des groupes de malades dans le temps afin de déterminer les caractéristiques du cours naturel d’une maladie. Cette connaissance de sa progression en l’absence de traitement peut servir ainsi à démontrer l’efficacité d’une thérapie potentielle lors d’un essai clinique. En France, une étude d’histoire naturelle (NatHis LAMA2), soutenue par l’AFM-Téléthon, a débuté en 2024 chez des enfants atteints de DMC liée à LAMA2. Cette première étude prospective française dans cette population a pour objectif de déterminer les caractéristiques de la maladie et son évolution au cours du temps.
En savoir plus :
- NatHis LAMA2 : le pourquoi du comment.
- Présentation du projet et des modalités de participation, avec sa coordinatrice Dr Andreea Seferian
- Les bases de données médicales (entrepôts de données de santé, registres…) recueillent et centralisent des informations sur des patients (maladies, caractéristiques personnelles...) sur une même plateforme.
Ce sont des outils indispensables pour mieux connaitre les maladies, surtout si elles sont rares, améliorer leur diagnostic et leur prise en charge, et faciliter l’identification de personnes pouvant être incluses dans les essais cliniques. Par exemple, le registre international des maladies musculaires congénitales, le CMDIR (Congenital Muscle Disease International Registry), recense des personnes atteintes d’une maladie congénitale du muscle, incluant les DMC.
En savoir plus : Un entrepôt de données de santé, c’est quoi ?


