Syndromes myasthéniques congénitaux
Les différents syndromes myasthéniques congénitaux sont tous liés à un dysfonctionnement, d’origine génétique, de la jonction entre les nerfs et les muscles. Ces maladies neuromusculaires rares et héréditaires sont traitables. Différents médicaments ont déjà transformé le quotidien de nombreux malades ! Des progrès restent toutefois à réaliser pour identifier l’ensemble des gènes responsables de ces maladies et améliorer encore leur traitement.
Quelle est leur cause ?
Les syndromes myasthéniques congénitaux (SMC) sont des maladies neuromusculaires dues à des anomalies de l'ADN, héréditaires.
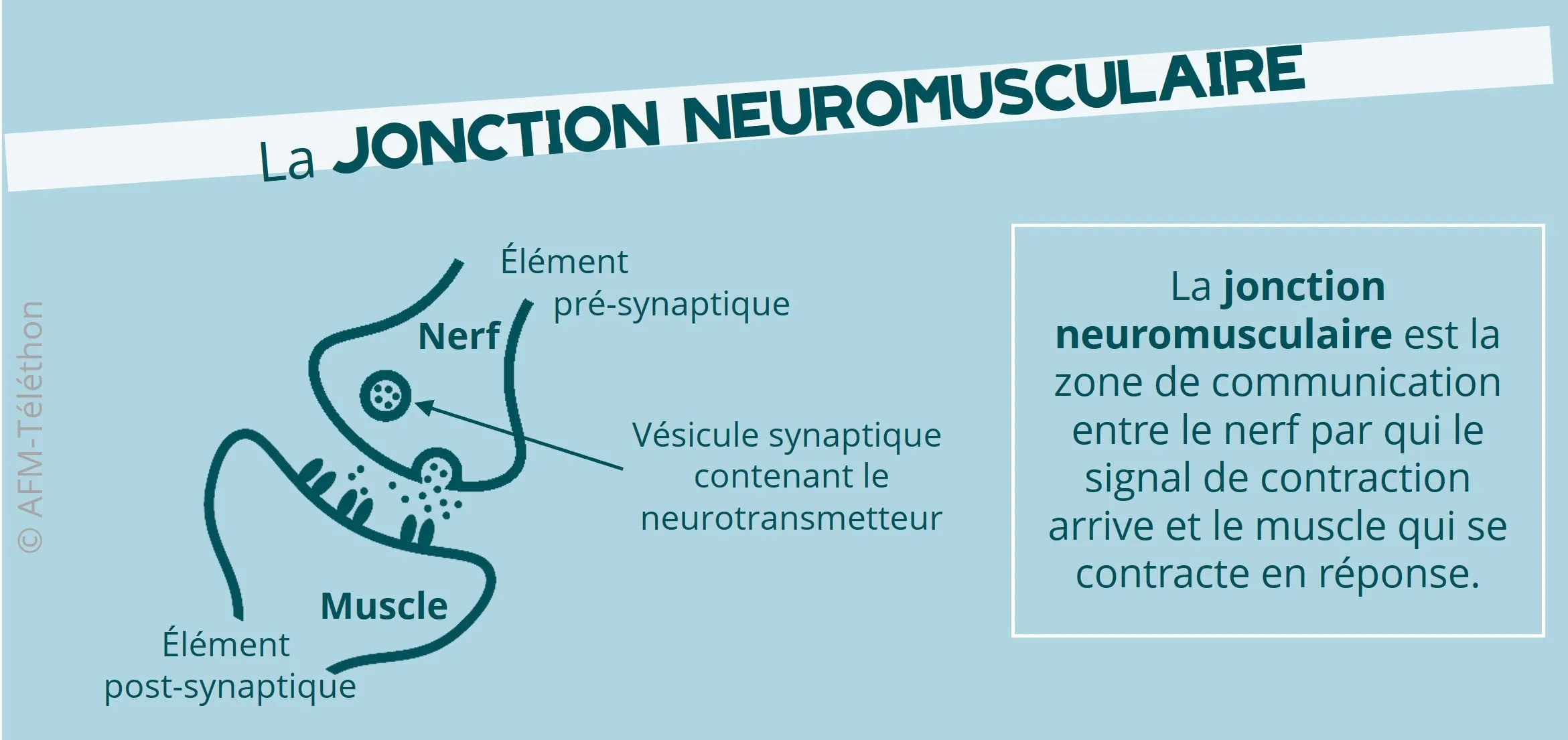
Quarante gènes sont désormais connus pour provoquer, lorsqu’ils présentent une mutation, un SMC. Le premier a été identifié en 1 995. Chacun de ces gènes code une protéine présente au niveau de la jonction neuromusculaire, c'est-à-dire de la zone de contact (synapse) entre le nerf et le muscle. Dans un syndrome myasthénique congénital, les anomalies de la protéine touchée entrainent un défaut de transmission de l’influx nerveux à l’origine des manifestations de la maladie.
Selon l'élément de la jonction neuromusculaire perturbé, on distingue les syndromes myasthéniques congénitaux :
- pré-synaptiques (en amont de la synapse, à l'extrémité du nerf),
- synaptiques (dans l'espace entre le nerf et le muscle),
- postsynaptiques (en aval de la synapse, au niveau de la membrane du muscle),
- avec un déficit de la glycosylation, liés à des gènes codant des enzymes impliquées dans l’ajout de sucre (glycosylation) sur certaines protéines de la jonction neuromusculaire, par exemple sur le récepteur de l'acétylcholine.
Une transmission familiale variable
L’anomalie génétique responsable d’un SMC se transmet à la descendance selon un mode le plus souvent autosomique récessif, beaucoup plus rarement autosomique dominant.
Les deux types de transmission sont possibles pour les gènes CHRNE et SYT2, selon la mutation en cause.
Un air de famille… ou pas !
Plusieurs membres d’une même famille peuvent être atteints par un syndrome myasthénique congénital donné, mais ce n’est pas toujours le cas. Il existe aussi des cas dits « sporadiques », où une seule personne de la famille est concernée.
Bon à savoir
- Les syndromes myasthéniques congénitaux débutent le plus souvent avant l’âge de 2 ans, et bien souvent dès les premiers jours de vie. Leur survenue à l’âge adulte est possible mais exceptionnelle.
- Certains symptômes, mêmes sévères, du tout-petit peuvent s’améliorer voire disparaitre ultérieurement de façon spontanée ou sous l’effet d’un traitement.
D’un muscle à l’autre
La faiblesse musculaire des SMC prédomine, dans la plupart des cas, au niveau des muscles de la tête et du cou entrainant une vision double (diplopie), des difficultés pour bouger les yeux (ophtalmoplégie ou ophtalmoparésie), garder les paupières ouvertes (ptosis), avoir des expressions marquées du visage (parésie ou paralysie facile) ou avaler (dysphagie). Les muscles respiratoires, ceux des membres et ceux du tronc (axiaux) peuvent également être touchés.
D'un âge de début à l'autre
L’intensité des symptômes et leur évolution sont très variables selon les personnes, le type de syndrome myasthénique congénital et l’âge auquel il commence à se manifester.
- À la naissance, la maladie peut s’exprimer par une faiblesse musculaire généralisée importante (hypotonie, se traduisant par une absence de mouvement spontané du nouveau-né), des difficultés pour respirer, téter ou avaler, des modifications de la voix ou des accès de défaillance respiratoire brutale.
- En cas de SMC apparu plus tard dans l’enfance, ce sont plutôt un retard d’acquisition de la station assise et/ou de la marche, une tête tombante, des difficultés à suivre un objet des yeux sans bouger la tête, un « strabisme », une fermeture des paupières et/ou des difficultés à s’alimenter qui attirent l’attention.
Ne pas confondre
• Les syndromes myasthéniques congénitaux sont aussi appelés « myasthénie infantile familiale ». Cette appellation peut porter à confusion avec une autre maladie : la myasthénie auto-immune.
• Les SMC et la myasthénie ont pour point commun de résulter d’un dysfonctionnement de la jonction neuromusculaire, soit la zone de communication entre le nerf (qui commande le mouvement) et le muscle.
• L’origine de ce dysfonctionnement est génétique pour les SMC. Elle est immunitaire pour la myasthénie auto-immune.
• Une personne atteinte de SMC peut recevoir, à tort, le diagnostic de myasthénie auto-immune. Dans ce cas l’existence d’une scoliose (chez l’enfant), l’inefficacité des médicaments contre la myasthénie et l’absence d’auto-anticorps caractéristique de cette maladie doivent faire remettre en cause le diagnostic et évoquer un SMC.
Comment est fait le diagnostic ?
Les syndromes myasthéniques congénitaux sont des maladies rares. En France, un peu moins de 600 personnes sont suivies en centres experts dans les maladies neuromusculaires pour un SMC. Et ces maladies sont qui plus est de découverte relativement récente : il a fallu attendre 1 977 pour que la première description d’un SMC soit publiée.
De ce fait, ces maladies restent mal connues des médecins non experts en maladies neuromusculaires, d’où un fréquent retard diagnostique. Selon une étude française de grande ampleur, le diagnostic de SMC n'est posé qu'à l'âge adulte dans près de six cas sur 10.
Deux facteurs de progrès en France
1. Les consultations spécialisées en maladies neuromusculaires sont à même de poser le diagnostic de syndrome myasthénique congénital.
2. Un Protocole national de diagnostic et de soins (PNDS) est disponible depuis 2021. Rédigé par des spécialistes des SMC, ce document décrit les manifestations, les examens à réaliser pour le diagnostic et la prise en charge optimale de ces maladies. Il s’accompagne d’un « synthèse à destination du médecin traitant ».
Pour affirmer qu’il s’agit d’un SMC, le médecin se base sur les manifestations de la maladie, l’existence éventuelle de cas comparables dans la famille, un examen clinique à la recherche de signes évocateurs, les résultats de l’électroneuromyogramme, l’absence d’auto-anticorps spécifiques de la myasthénie auto-immune (anti-RACh, anti-MuSK) et la présence d'anomalies dans un gène connu pour donner un SMC, identifiées par un test génétique et qui permet d’affirmer avec certitude le diagnostic. L’adoption des nouvelles méthodes de séquençage, à haut débit, ou next-generation sequencing (NGS) devrait réduire les délais diagnostiques.
Attention néanmoins, les chercheurs n’ont pas encore identifié tous les gènes responsables de ces maladies. L’analyse génétique peut donc ne révéler aucune anomalie alors qu’il s’agit bien d’un SMC : c’est le cas dans près d'un cas sur trois.
Quels sont les médicaments disponibles ?
Selon le gène en cause et l’importance des symptômes, le médecin peut prescrire un ou plusieurs médicaments : anticholinestérasiques (Prostigmine®, Mestinon®, Mytelase®…), éphédrine, salbutamol, 3,4-diaminopyridine ou amifampridine (Firdapse®), fluoxétine…
Bon à savoir
- L’efficacité de chaque médicament est variable selon le SMC en cause et parfois également selon les personnes atteintes du même SMC.
- Un médicament efficace dans un type SMC peut s’avérer inefficace voire délétère dans un autre type.
- Les médicaments sont parfois associés entre eux.
- Un SMC n’entrainant qu’une légère gêne peut ne pas nécessiter de médicament spécifique.
Il s’agit de médicaments « repositionnés » dans les syndromes myasthéniques congénitaux : ils sont utilisés depuis des années pour soigner d’autres maladies (asthme, troubles du rythme cardiaque…).
Même si leurs mécanismes d'action ne sont pas toujours très bien compris, ils ont modifié du tout au tout le traitement et le pronostic des SMC. Ils ne font pas disparaître la maladie (l’anomalie génétique persiste) mais peuvent entrainer une amélioration très importante des symptômes, y compris lorsqu’ils sont sévères avec par exemple une possible reprise de la marche alors que la faiblesse musculaire obligeait, avant le traitement, à utiliser un fauteuil roulant.
En complément des médicaments
Afin de réduire la faiblesse musculaire et de prévenir les complications, la prise en charge peut comporter au cas par cas, outre les médicaments et selon l’importance des symptômes :
- de la kinésithérapie pour entretenir la musculature, mais aussi pour limiter les rétractions musculaires et conserver l’amplitude des articulations, parfois en association avec le port d’attelles et la chirurgie,
- des séances d’orthoptie (rééducation des yeux) et d’orthophonie (voix et troubles de la déglutition),
- des séances de kinésithérapie respiratoire et une assistance à la ventilation (ventilation non invasive ou par trachéotomie si nécessaire),
- le conseil génétique, pour s’informer sur le mode de transmission de la maladie et sur le risque qu’une personne a de développer et/ou de transmettre la maladie dans l'avenir,
- un suivi en ergothérapie avec si nécessaire la mise en place d’aides techniques (pour les difficultés d’écriture notamment),
- une prise en charge par un neuropsychologue le cas échéant,
- une prise en charge nutritionnelle (compléments alimentaires, nutrition par sonde…),
- une activité physique adaptée, régulière et modérée, encadrée par un professionnel.
Qui consulter pour le suivi et à quelle fréquence ?
- Dans l’idéal, une personne atteinte d’un syndrome myasthénique congénital devrait être suivie par une équipe dite « pluridisciplinaire » c’est-à-dire rassemblant différents professionnels de santé et coordonnée par un neuropédiatre ou neurologue, dans un Centre de référence ou de compétences dédié aux maladies neuromusculaires, en lien avec son médecin traitant.
- Une consultation pluridisciplinaire spécialisée devrait être planifiée au moins deux fois par an, pour notamment évaluer la croissance et le développement chez l’enfant, la gêne musculaire, le déficit moteur et la tolérance du traitement, vérifier les vaccinations et réaliser différents examens (prise de sang, radiographie, épreuves fonctionnels respiratoires…).
Des précautions avec l’automédication
Certains médicaments sont formellement contre-indiqués et d’autres à utiliser avec précautions quand on a un syndrome myasthénique congénital. En cas de doute, n’hésitez pas à demander conseil à votre médecin ou à votre pharmacien.
| Famille de médicaments | Formellement contre-indiqués | A utiliser avec précaution |
|---|---|---|
| Antibiotiques | Aminosides en injection, Colistine, Cyclines injectables, Télithromycine | Aminosides et polymines en application locale, Lincomycine, Clindamycine, Fluoroquinolones |
| Cardiovasculaires | Quinidine, Bêtabloquants, Procaïnamide | Lidocaïne en intraveineux |
| Anesthésiques | Curarisants | Anesthésiques volatils, Barbituriques en intramusculaire ou intraveineux, Kétamine, Propanidide |
| Neurologiques | Triméthadione, Diphénylhydantoïne, Dantrolène | Carbamazépine, Chlorpromazine, Lithium |
| Divers | D-pénicillamine, Magnésium en intraveineux, Quinine et Chrloroquinine, Halofantrine, Méfloquine, Bêtabloquants en collyre, Oxybutynine | Benzodiazépines, Phénothiazine, Magnésium per os, Interféron alpha, Patch à la nicotine |
(Source : Orphanet Urgences)
Y a-t-il des travaux de recherche ?
Ces cinq dernières années ont été publiés plus de 300 articles scientifiques et médicaux sur les syndromes myasthéniques congénitaux, témoignant de l’intérêt des chercheurs et médecins pour ces maladies. Leur travaux permettent de mieux connaitre les SMC (nouveaux gènes, nouveaux variants, manifestations, évolution…) mais aussi de mieux les soigner.
Différentes manières nouvelles de traiter les syndromes myasthéniques congénitaux sont ainsi à l’étude. La thérapie génique en fait partie, avec par exemple des travaux précliniques (souris) portant sur des virus adénoassociés (AAV) apportant le gène COLQ ou le gène DOK7, dans des modèles de SMC liés respectivement à COLQ ou à DOK7.
Un essai clinique pour l’ARGX-119
Depuis septembre 2024, un essai évalue la sécurité, la tolérance, le devenir dans l’organisme et l’efficacité d’un médicament en développement, l’ARGX-119, contre placebo. Il se déroule dans plusieurs pays dont la France qui compte deux sites investigateurs, Marseille et Paris. Ses 16 participants sont des adultes atteints d’un SMC lié à DOK7.
L’ARGX-119 active de façon spécifique la protéine MuSK, ou tyrosine-kinase spécifique du muscle, avec l’objectif de stabiliser la jonction neuromusculaire. Ses effets chez un modèle murin de la maladie sont encourageants.
À noter que ce candidat-médicament est également en phase d’essai clinique dans la maladie de Charcot (sclérose latérale amyotrophique) et d’exploration préclinique dans la myasthénie auto-immune.
Une étude d’histoire naturelle en France
Ce que l’on appelle « histoire naturelle » d’une maladie correspond à la description de ses différentes manifestations et de leur évolution au cours du temps. Sa connaissance est essentielle notamment pour pouvoir juger des bénéfices d’un nouveau médicament. Une telle étude est en cours depuis février 2024 dans plusieurs pays dont la France (à Bordeaux, Lille, Marseille et Paris) chez 100 participants, âgés de deux ans et plus, atteints d’un syndrome myasthénique congénital lié à DOK7, MUSK, AGRN ou LRP4.
Les gènes, une vaste sujet
Au-delà d’identifier de nouveaux gènes, il s’agit de comprendre quelles sont les conséquences précises de leur mutation. En 2023, une équipe française impliquant l’un des laboratoires de l’AFM-Téléthon, I-Stem, a par exemple contribué à mieux comprendre les conséquences fonctionnelles d'une mutation affectant l'expression du gène ColQ.
Un groupe dédié en France
Des médecins et des chercheurs de l'Institut de Myologie (Paris) ont créé en 2001 le réseau français des Syndromes myasthéniques congénitaux. Il regroupe des experts médico-scientifiques travaillant sur les syndromes myasthéniques congénitaux. Leur dernière réunion annuelle a eu lieu en février 2025.



Comment se manifestent-ils ?
Les différents syndromes myasthéniques congénitaux provoquent une fatigabilité excessive et une faiblesse musculaire qui tend à s’aggraver à l’effort et fluctue souvent au fil des heures, augmentant en fin de journée.
Ces symptômes peuvent également s’exacerber à l’occasion d’une infection, d’une fièvre ou d’un stress prolongé. En dehors de ces circonstances particulières les manifestations d’un SMC restent stables, dans la majorité des cas.