Myopathies des ceintures
Les myopathies des ceintures se manifestent de façons diverses allant d’une simple fatigabilité à des formes entraînant la perte de la marche, avec ou sans complications cardiaque et/ou respiratoire. Un suivi médical régulier permet de traiter à temps et limiter les conséquences de la maladie. Les thérapies innovantes sont des pistes de recherche en plein essor dans ces maladies.
Qu’est-ce que c’est ?
Les myopathies des ceintures, ou LGMD (pour Limb-Girdle Muscular Dystrophy) sont des maladies génétiques rares qui touchent principalement les muscles (on parle de myopathie) dits « proximaux », c’est-à-dire ceux des épaules (ceinture scapulaire) et des hanches (ceinture pelvienne), qui diminuent progressivement de volume, s’affaiblissent et se régénèrent difficilement.
Elles apparaissent souvent avant l’âge de 20 ans, mais peuvent aussi débuter plus tardivement (dans de rares cas au-delà de 70 ans). Les premiers signes sont principalement des difficultés pour courir, pour monter les escaliers et se relever du sol. Les chutes sont fréquentes. Paradoxalement, les mollets peuvent paraître très musclés sans pour autant être particulièrement forts (on parle de « pseudo-hypertrophie »).
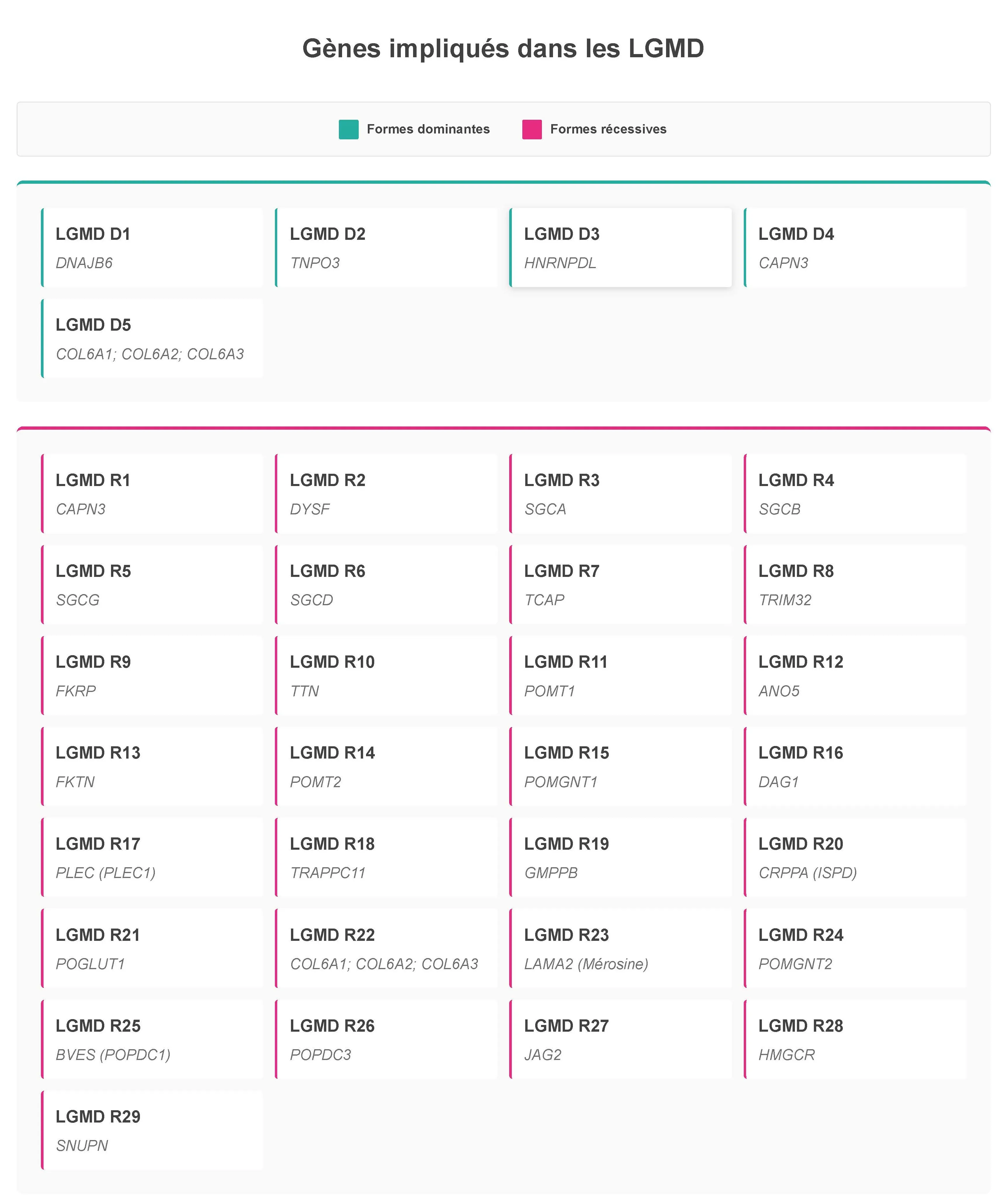
Trente-quatre formes de LGMD ont été répertoriées à ce jour. On y distingue souvent six groupes (en fonction des mécanismes menant à la maladie) :
• Anoctaminopathies : LGMD R12
• Calpaïnopathies : LGMD R1 et D4
• Collagénopathies : LGMD R22 et D5
• Dysferlinopathies : LGMD R2
• Dystroglycanopathies : LGMD R9, R11, R13, R14, R15, R16, R19, R20 et R24
• Sarcoglycanopathies : LGMD R3, R4, R5 et R6
Combien de personnes sont concernées ?
Il est estimé qu’environ 1 à 3 personnes sur 100 000 sont concernées, toutes formes de LGMD confondues. La banque nationale de données maladies rares (BNDMR) recense environ 2 400 cas en France. La LGMD R1 (calpaïnopathie) est la fréquente (30 % des cas de LGMD en France), suivie de la LGMD R2 (dysferlinopathie), la LGMD R9, la LGMD R12 (anoctaminopathie) et, collectivement, des LGMD R3 à R6 (sarcoglycanopathies).
Dans certaines petites populations, la fréquence d’une ou plusieurs LGMD est particulièrement élevée, comme la LGMD R1 chez les Petits Blancs des Hauts (la Réunion), les Basques ou les amish (États-Unis) ; la LGMD R2 dans certaines parties du Maghreb, du Moyen-Orient ou de l’Inde ; la LGMD R5 chez les amish et les populations tziganes…
Focus sur les formes les plus fréquentes : quelques spécificités…
Que peut-on faire ?
La prise en charge médicale des LGMD est pour l’instant uniquement symptomatique ; elle vise essentiellement à prévenir les complications possibles (rétractions, diminution de la force musculaire, baisse des capacités pulmonaires…). Les recherches continuent pour trouver un traitement dit « de fond », qui cible la cause de la maladie, avec des pistes thérapeutiques prometteuses et innovantes à l’étude.
VOIR LES AVANCÉES 2026 DANS LES MYOPATHIES DES CEINTURES
Pour une prise en charge optimale, les patients atteints de myopathies des ceintures doivent être suivis par une équipe médicale au sein de consultations pluridisciplinaires spécialisées dans les maladies neuromusculaires.
- Une surveillance annuelle est recommandée pour faire un bilan orthopédique (muscles, tendons, ligaments et os), cardiaque et respiratoire.
- La prise en charge orthopédique (kinésithérapie, appareillage) doit être précoce, régulière et adaptée à chaque situation individuelle. Elle permet notamment de maintenir autant que possible la souplesse des muscles et des articulations.
- Des aides techniques (cannes, supports de bras…) peuvent compenser certaines faiblesses musculaires et permettre de continuer à réaliser des gestes de la vie quotidienne devenus difficiles ou impossibles avec l’évolution de la maladie. Les fauteuils roulants et scooters électriques font partie des aides techniques à la mobilité et permettent de retrouver de l’autonomie dans ses déplacements.
- L’atteinte des muscles respiratoires, nécessite une prise en charge spécifique et individualisée.
- Un suivi cardiologique régulier est nécessaire pour dépister au plus tôt une atteinte cardiaque éventuelle et mettre en place rapidement un traitement (médicaments, pacemaker, défibrillateur).
- Le conseil génétique se réalise en consultation avec un expert en génétique qui informe une personne, ou une famille, sur les risques de développer ou de transmettre la maladie. Il est une ressource importante dans le cadre d’un projet de grossesse.
La carte d’urgence LGMD, mise en place par la filière FILNEMUS, présente les recommandations importantes pour la sécurité et la prise en charge adéquate d’une personne atteinte de LGMD en cas d’urgence.
LGMD R1 ou LGMD 2A ?
Depuis la publication en 2018 de travaux du European Neuromuscular Centre (ENMC), les communautés médicale et scientifique utilisent pour les LGMD une dénomination et une classification mises à jour. Le nom des différentes formes de LGMD prend dès lors le format suivant : LGMD « D » ou « R » pour le mode de transmission (dominant ou récessif), suivi d’un chiffre, indiquant l’ordre de découverte, et du nom de la protéine en cause. Ainsi la « LGMD 2A » devient la « LGMD R1 liée à la calpaïne 3 » (souvent raccourci en « LGMD R1 »). Toutefois, les anciennes appellations sont encore quelques fois utilisées dans la littérature médico-scientifique ou par la communauté LGMD.
À quoi sont-elles dues ?
Les LGMD sont dues à des anomalies pathologiques présentes dans des gènes indispensables au bon fonctionnement des muscles. Depuis la découverte du gène de la calpaïne 3 en 1995 par l’équipe d’Isabelle Richard à Généthon, 34 gènes différents ont été identifiés dont certaines mutations peuvent causer des LGMD. Héréditaires, les LGMD se transmettent principalement de manière autosomique récessive (LGMD « R » pour récessives, 29 formes décrites représentant 90 % des LGMD), et plus rarement sur le mode autosomique dominant (LGMD « D » pour dominantes, cinq formes). Dans certains cas, elles peuvent être dues à des mutations dites « de novo », qui n’étaient présentes chez aucun des parents.
Comment fait-on le diagnostic ?
Il se base sur l’identification des muscles atteints (ceux des ceintures), l’évolution de la maladie, l’histoire familiale (transmission génétique) et, pour un diagnostic de certitude, un test génétique révélant une ou des mutations pathologiques dans un des gènes impliqués (connus à ce jour) dans les LGMD. Le diagnostic peut être complété par d’autres examens, tels que prise de sang, imagerie des muscles, électromyogramme, biopsie musculaire… La prise de sang permet de recueillir des cellules (notamment des globules blancs) pour les analyses génétiques, et de mesurer la concentration sanguine de l’enzyme musculaire créatine kinase (CPK). Le fragment de muscle prélevé par biopsie musculaire permet de rechercher des altérations du tissu musculaire caractéristiques des LGMD (hétérogénéité de tailles des fibres musculaires, infiltrations graisseuses, fibrose…) et d’identifier, dans certains cas, la protéine déficitaire et/ou dysfonctionnelle responsable de la maladie.
Les essais cliniques : à la recherche de traitements
Plusieurs stratégies thérapeutiques ciblant la cause de la maladie sont à l’étude afin d’agir sur les différents mécanismes pathologiques propres à chaque forme de LGMD.
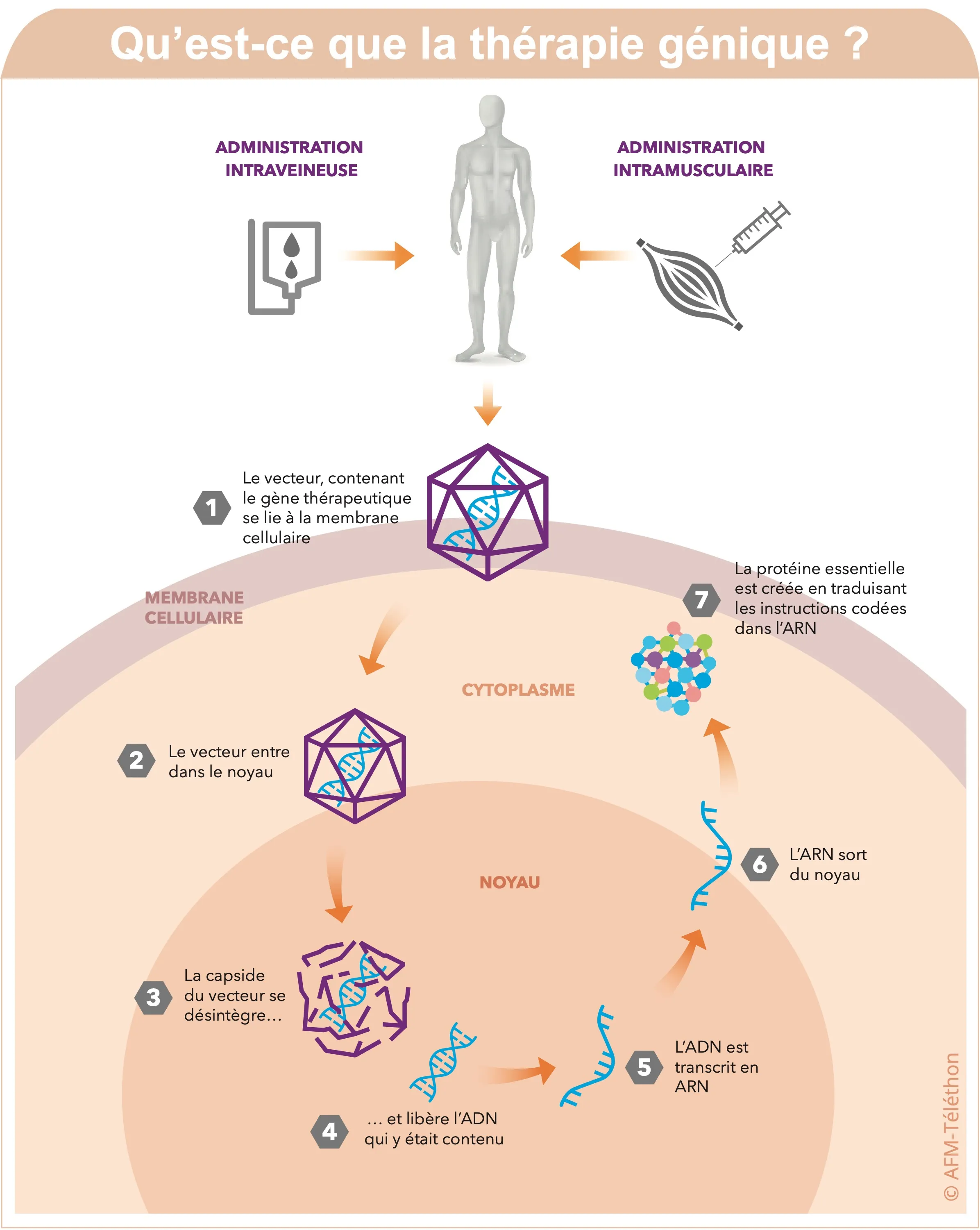
La thérapie génique
La thérapie génique consiste à insérer dans les cellules de la personne malade un « gène-médicament » afin d’apporter une copie fonctionnelle du gène déficient en cause dans la maladie. Plusieurs essais cliniques de thérapie génique sont en cours dans les LGMD, avec des résultats encourageants pour certains programmes.
- Dans la LGMD R4 liée à SGCB (sarcoglycanopathie), l'essai de phase III EMERGENE du produit de thérapie génique SRP-9003 (Sarepta Therapeutics) a montré des résultats encourageants : à 60 jours, la protéine bêta-sarcoglycane est restaurée à des niveaux cliniquement pertinents et des signes de réduction de la dégénérescence musculaire sont observés. Cependant, des effets indésirables hépatiques ont été identifiés chez une majorité de participants de l'essai, contribuant à la suspension clinique imposée par la FDA en juillet 2025 sur l'ensemble des programmes de Sarepta, dont EMERGENE, utilisant le vecteur AAVrh74. La trajectoire réglementaire du SRP-9003 reste à ce jour incertaine.
- Dans la LGMD R5 liée à SGCG (sarcoglycanopathie), Atamyo Therapeutics, une société de biotechnologie créée en 2020 par Généthon sur la base des travaux de recherche de l'équipe d'Isabelle Richard, chercheuse au Généthon, développe le produit de thérapie génique ATA-200 en partenariat avec l'Université de Floride. Quatre enfants ont été traités aux États-Unis à ce jour dans l'essai ATA-003-GSAR. Les résultats sont encourageants : chez les deux premiers patients suivis sur un an, plus de 90 % des fibres musculaires examinées expriment la protéine manquante, avec des améliorations fonctionnelles et aucun effet secondaire grave rapporté. La première cohorte américaine est terminée ; le volet européen de l'essai n'a pas encore démarré.
- Dans la LGMD R9 liée à FKRP, Atamyo Therapeutics a démarré en 2022 l'essai clinique ATA-001-FKRP international (incluant la France) pour tester le produit de thérapie génique ATA-100. La phase de détermination de dose, menée chez six participants, est terminée depuis avril 2025. À un an de suivi, les participants traités respirent légèrement mieux, marchent plus vite et voient leurs capacités motrices globales progresser. La prochaine étape consiste à inclure de nouveaux patients par une extension de cohorte, en attendant le lancement d'un essai pivot destiné à confirmer l'efficacité du produit.
Aux États-Unis, Asklepios BioPharmaceutical (AskBio) évalue l'efficacité d'un autre gène-médicament pour la LGMD R9, le LION-101, dans l'essai LION-CS101. Les données de sécurité de la première cohorte (5 patients), avec un an de recul, sont rassurantes : aucun effet indésirable lié à la dose ni aucun événement grave n'a été rapporté ; de légères élévations des enzymes du foie observées après l'injection se sont normalisées spontanément. Le recrutement de la deuxième cohorte est en cours.
La thérapie pharmacologique
Elle utilise des petites molécules de natures différentes (sucres, protéines, acides aminés…) pour corriger des processus biologiques (glycosylation, autophagie…) perturbés chez le malade.
- Dans la LGMD R9 liée à FKRP, le ribitol (BBP-418), pris par voie orale, est une molécule candidate développée par BridgeBio Pharma, évaluée chez 112 personnes dans l'essai de phase III FORTIFY. L'analyse des patients après un an de suivi montre une amélioration de la fonction motrice, de la vitesse de marche et de la capacité respiratoire, avec une bonne tolérance du produit. BridgeBio a déposé une demande d'autorisation de mise sur le marché (NDA) auprès de la FDA américaine au premier trimestre 2026. Une décision est attendue pour le dernier trimestre 2026. Si elle est approuvée, le ribitol deviendrait le premier traitement spécifique d'une myopathie des ceintures disponible sur le marché. En Europe, des démarches pour une autorisation accélérée sont en cours de discussion.
Depuis 2015, le laboratoire I-stem travaille sur des cellules modèles de myopathies des ceintures pour identifier des candidats-médicaments parmi des molécules déjà connues.
Les connaissances progressent
Les recherches ont montré que certains des gènes en cause dans les LGMD sont aussi impliqués dans d’autres formes de maladies neuromusculaires, notamment dans plusieurs formes de dystrophies musculaires congénitales, de myopathies distales ou de myopathies myofibrillaires.
Des études observationnelles et des bases de données sont mises en place pour mieux décrire les différentes formes de LGMD et leur évolution. Ces données sont particulièrement précieuses pour la préparation de futurs essais cliniques.
En France, trois de ces études sont en cours sur les LGMD :
- L’étude CALNATHIS évalue sur deux ans la perte de force des bras et des jambes chez 25 patients atteints de LGMD R1 liée à CAPN3.
- Le registre C3R, soutenu par l’AFM-Téléthon, est dédié aux calpaïnopathies, et recense des données médicales et génétiques issues du suivi, en consultation neuromusculaire de la filière Filnemus, d’enfants et d’adultes atteints de ces maladies en France.
- Le registre SARCO, développé également par l’AFM-Téléthon, rassemble des informations telles que l’âge de début de la maladie et ses manifestations, le gène en cause, le type de mutation, les résultats de certaines prises de sang, d’évaluations de la force musculaire des patients atteints de sarcoglycanopathies en France.
Le groupe d’intérêt AFM-Téléthon LGMD
Il rassemble des malades atteints de LGMD, experts de cette maladie. En plus du partage d’expériences, il apporte une information médicale et scientifique régulière sur son blog ou lors de journées d’information, en lien avec des médecins et chercheurs impliqués dans les LGMD.
https://lgmd.afm-telethon.fr/blog/




Comment évoluent-elles ?
Leur évolution est très variable, y compris au sein d’une même famille de malades, mais est en général relativement plutôt lente. Les manifestations cliniques sont le plus souvent limitées aux muscles du mouvement, aussi appelés muscles squelettiques. Selon le type de LGMD, les muscles de la respiration (difficultés respiratoires) et/ou du cœur (cardiomyopathie, troubles du rythme) peuvent être atteints.
La perte de la force musculaire et les changements de structure (dystrophie) des muscles provoquent leur enraidissement et celui des articulations (on parle de rétractions) qui peut entraîner des déformations articulaires (scoliose, colonne raide…).
Avec le temps, la marche peut devenir difficile, voire impossible dans certaines formes (comme au sein des sarcoglycanopathies), tandis que d’autres sont assez peu ou pas évolutives, et les malades peuvent conserver la marche (comme dans les anoctaminopathies).