Amyotrophie spinale proximale liée à SMN1
Depuis quelques années, l’amyotrophie spinale proximale liée à SMN1 (SMA) connait une véritable révolution thérapeutique avec l’arrivée de trois traitements : le Spinraza®, le Zolgensma® et l’Evrysdi®. Plus ils sont administrés tôt, plus ils sont efficaces. Ils doivent être associés à une prise en charge symptomatique de cette maladie due à une dégénérescence des motoneurones, qui se manifeste par une faiblesse musculaire.
Quelle est la cause de l'amyotrophie spinale ?
L'amyotrophie spinale proximale liée à SMN1 (SMA ou amyotrophie spinale infantile, ASI) est due à des anomalies situées dans le gène SMN1, localisé sur le chromosome 5.
Ces anomalies entrainent l’absence de production de la protéine de survie des motoneurones SMN (pour survival of motor neuron), avec une perte de motoneurones, des cellules nerveuses qui commandent la contraction des muscles. Cela se traduit par une faiblesse et une fonte des muscles (amyotrophie) dit « proximaux », c’est-à-dire les plus proches du tronc : muscles des épaules et des bras pour les membres supérieurs et muscles des hanches et des cuisses pour les membres inférieurs.

Chez toutes les personnes atteintes de SMA, et chez 95% de la population générale, il existe en plus du gène SMN1 un autre gène quasiment identique, le gène SMN2. Cependant, ce gène SMN2 ne suffit pas à produire suffisamment de protéine SMN fonctionnelle.
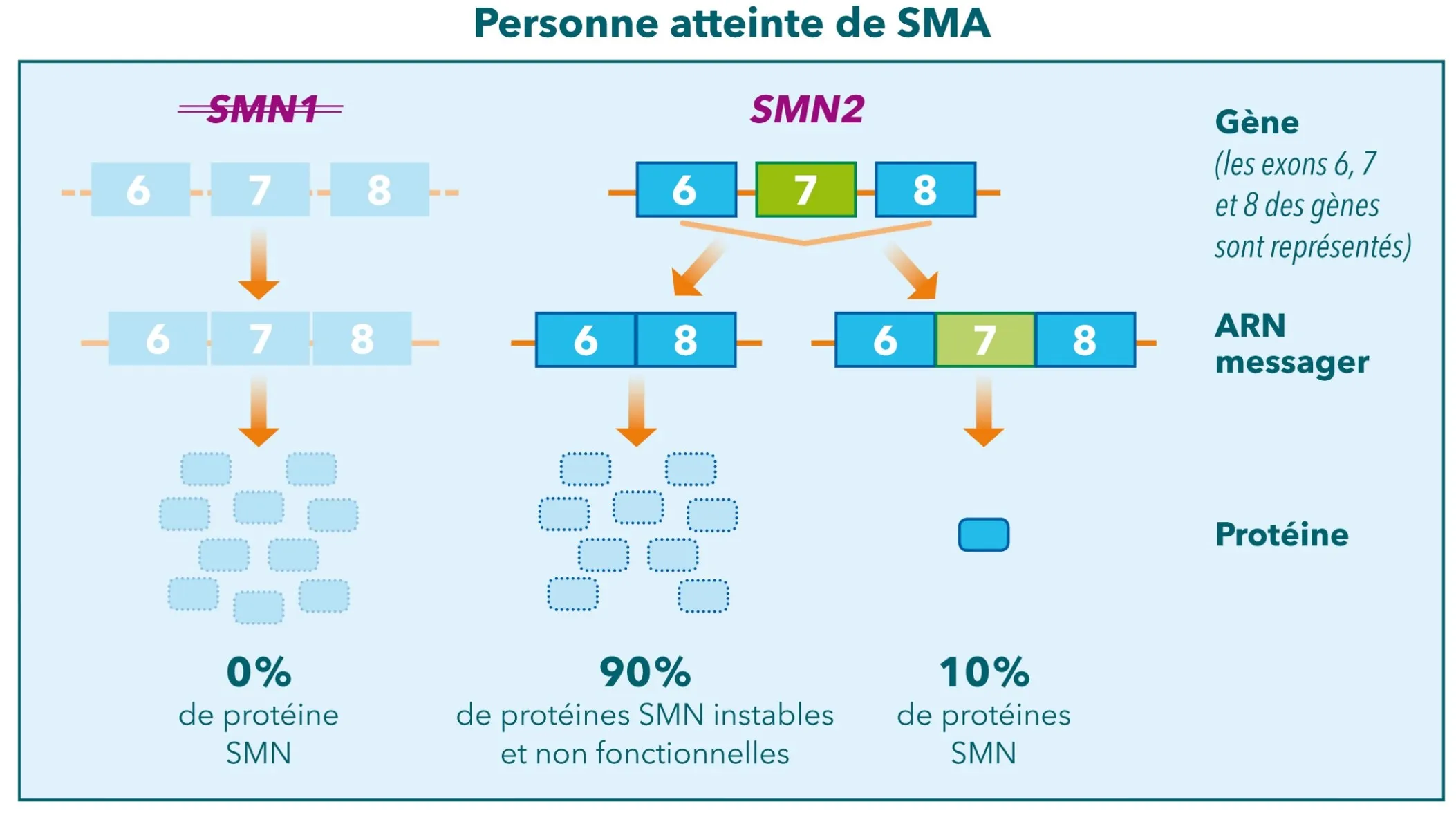
Les médicaments actuels et ceux à l’étude dans la SMA ciblent principalement les gènes SMN1 ou SMN2 dans le but d’augmenter la production de protéine SMN.
Les différents noms de la SMA
L'amyotrophie spinale proximale liée à SMN1 est aussi appelée amyotrophie spinale infantile (ASI), amyotrophie spinale 5q, maladie de la corne antérieure de la moelle épinière, maladie du motoneurone, amyotrophie spinale antérieure (ASA), ou, plus fréquemment, SMA (pour spinal muscular atrophy).
Il existe des formes d'amyotrophie spinale proximale liées à d'autres gènes, mais l'amyotrophie spinale proximale liée à SMN1 (SMA) est la forme la plus fréquente.
L’AFM vous accompagne
- via ses Services régionaux et leurs Référents parcours de santé (RPS) : pour trouver des solutions adaptées à votre situation (suivis médicaux, recherche de consultations spécialisées de proximité, démarches administratives, acquisition de matériel…).
- via son Groupe d’Intérêt Amyotrophies Spinales, composé de personnes concernées par la SMA : à l’écoute des malades, pour informer sur les amyotrophies spinales, organiser des rencontres ou des journées d’information…
- via ses Délégations départementales, regroupant des bénévoles concernés par la maladie : pour assurer un lien auprès des malades.
Vous souhaitez en savoir plus ou avez une question particulière ? La ligne Accueil Familles de l’AFM-Téléthon est à votre écoute au 0 800 35 36 37 (appel gratuit).
Dans quelles circonstances est posé le diagnostic ?
● À la naissance, en l’absence de symptômes
L’arrivée des nouveaux traitements et la démonstration de l’intérêt de les utiliser précocement pour modifier l’évolution de l’amyotrophie spinale proximale liée à SMN1 (SMA), voire stopper sa progression, a poussé à développer son dépistage à la naissance dans plusieurs pays.
Le dépistage à la naissance de la SMA
Aussi appelé dépistage néonatal (DNN), il permet de savoir dès les premiers jours de vie, et le plus souvent avant même l’apparition des premiers symptômes, si un nouveau-né est porteur ou non d’anomalies génétiques de la SMA. Depuis quelques années, plusieurs régions ou pays du monde ont démontré que le dépistage néonatal de la SMA est faisable et efficace.
En France, le dépistage systématique de la SMA chez tous les nouveau-nés est réalisé depuis le 1er septembre 2025.
Pour en savoir plus sur le dépistage néonatal de la SMA en France en pratique
Cela fait suite aux résultats très positifs du programme pilote DEPISMA, soutenu par l'AFM-Téléthon, lequel a démontré la faisabilité du dépistage néonatal de la SMA sur une durée de deux ans dans deux régions : la Nouvelle-Aquitaine et la région Grand Est.
Dépister au plus tôt pour traiter au plus tôt
Dans l’amyotrophie spinale infantile (SMA), les trois médicaments autorisés à ce jour, le Spinraza®, le Zolgensma® et l’Evrysdi®, ont montré une efficacité plus importante et plus rapide s’ils sont initiés avant les premiers symptômes (en présymptomatique) qu’après (en phase symptomatique). D’où l’importance de diagnostiquer précocement la maladie pour permettre aux nouveau-nés de bénéficier d’un traitement et d’une prise en charge adaptée immédiate, leur donnant ainsi les meilleures chances pour l’avenir.
Le nombre de copies du gène SMN2, un indicateur de la sévérité de la maladie ?
Le dépistage néonatal de la SMA repose dans un premier temps sur la recherche d’absence de gène SMN1 puis, si le test est positif, sur la détermination du nombre de copies du gène SMN2. Connaitre ce nombre permet de renseigner sur l’évolution possible de la SMA, notamment en l’absence de symptômes : plus le nombre de copies de SMN2 (entre 1 et 6) est élevé, plus il y a de protéine SMN fonctionnelle produite, moins la maladie est supposée sévère.
Ces informations aident les médecins, lors des Réunions de Consultations Pluridisciplinaires (RCP), à déterminer le traitement ou la prise en charge le plus adapté pour le bébé diagnostiqué positivement.
● Avant la naissance, lorsque la maladie existe dans la famille
D’origine génétique, l’amyotrophie spinale proximale liée à SMN1 (SMA) est une maladie héréditaire, qui se transmet selon un mode autosomique récessif.
Si un membre d’une famille est atteint de SMA, d’autres membres de cette même famille peuvent avoir un risque de l’être aussi. Le recours au conseil génétique permet d’évaluer ce risque et d'accompagner une personne ou une famille confrontée à ce risque ou lors d’un projet d’enfant.
Lors de la grossesse, le diagnostic prénatal (ou anténatal) permet de déterminer si l'enfant à naître est porteur ou non d'anomalies génétiques en cause dans l’amyotrophie spinale proximale liée à SMN1.
● Plus tardivement, en présence de symptômes
Aujourd’hui, le diagnostic de l’amyotrophie spinale proximale liée à SMN1 (SMA) est le plus souvent posé lors de l’apparition des premiers symptômes. Il s’agit principalement d’une faiblesse musculaire, d’importance variable, qui retentit sur les capacités motrices et la fonction respiratoire. Elle peut apparaître dès la naissance ou dans les tous premiers mois de vie, chez l'enfant, et plus rarement chez l'adolescent, voire à l’âge adulte.
La classification actuelle distingue quatre formes d'amyotrophie spinale proximale, selon l’âge de début de la maladie et la sévérité des symptômes. Il est généralement admis que plus les symptômes apparaissent tôt, plus l'évolution de la maladie est sévère.
- L'amyotrophie spinale de type I (SMA de type I ou maladie de Werdnig-Hoffmann) commence avant l'âge de 6 mois, parfois même dès la naissance. L’enfant n’est pas capable de s’assoir seul. Certains experts font aussi la distinction entre une forme très précoce de la maladie se manifestant avant la naissance par une diminution des mouvements du fœtus (forme dite type 0), la forme débutant entre la naissance et 3 mois (type I) et la forme débutant après 3 mois (type I bis). D’autres lui préfèrent une classification basée sur 3 lettres (A, B et C).
- L'amyotrophie spinale de type II (SMA de type II) a un début plus tardif, entre l'âge de 6 et 18 mois. L’enfant est capable de se tenir assis seul mais n'acquiert pas la marche.
- Dans l'amyotrophie spinale de type III (SMA de type III ou maladie de Kugelberg-Welander), les symptômes commencent après l'âge de 18 mois, généralement avant l’âge de 6 ans et évoluent lentement, sur de nombreuses années.
- Dans l'amyotrophie spinale de type IV (ou SMA de type IV), les symptômes débutent à l'âge adulte avec des difficultés à la marche. L’évolution est généralement très lente, voire absente dans certains cas.
Les termes « non assis/non sitter (en anglais) », « assis/sitter » ou « ambulant/walker » peuvent aussi être employés pour définir la prise en charge en fonction des capacités motrices acquises.
Depuis l’arrivée des traitements, de nouveaux profils d’enfants atteints de SMA sont observés et pourraient faire évoluer la classification de la maladie. En effet, l’administration d’un traitement (en particulier la thérapie génique) très tôt après la naissance permet d’agir sur la maladie, son développement et son évolution : la plupart des nouveau-nés traités avant l’apparition des symptômes ont un développement moteur favorable/normal (avec un recul de quelques années).
Il reste cependant encore des éléments à adapter, comme le suivi médical et la prise en charge au quotidien qui se poursuivent en prenant en compte la situation de l’enfant différente de celle d’un enfant du même âge avec une SMA, sans traitement. Les équipes médicales du réseau Filnemus construisent cette nouvelle prise en charge en s’appuyant sur leur expertise et l’expérience médicale internationale (publications, données en vie réelle…).
Voir l’actualité « SMA de type I : deux ans de thérapie génique, les résultats d’une étude française »
La sévérité de la maladie semble également être étroitement liée au nombre de copies du gène SMN2 :
Lorsqu’une amyotrophie spinale (SMA) est suspectée, le médecin propose des examens complémentaires, comme le test génétique qui confirmera ou non le diagnostic de la maladie. Dans la SMA, il s’agit principalement de rechercher l’absence de copies du gène SMN1, en cause dans la maladie, et de déterminer le nombre de copies du gène SMN2. Ainsi, on peut distinguer les types de SMA.

Déjà trois médicaments
Longtemps sans traitement de « fond », c’est-à-dire qui ciblent la cause et non les conséquences de la maladie, l’amyotrophie spinale proximale liée à SMN1 (SMA) dispose désormais de trois traitements : le Spinraza®, le Zolgensma® et l’Evrysdi®.
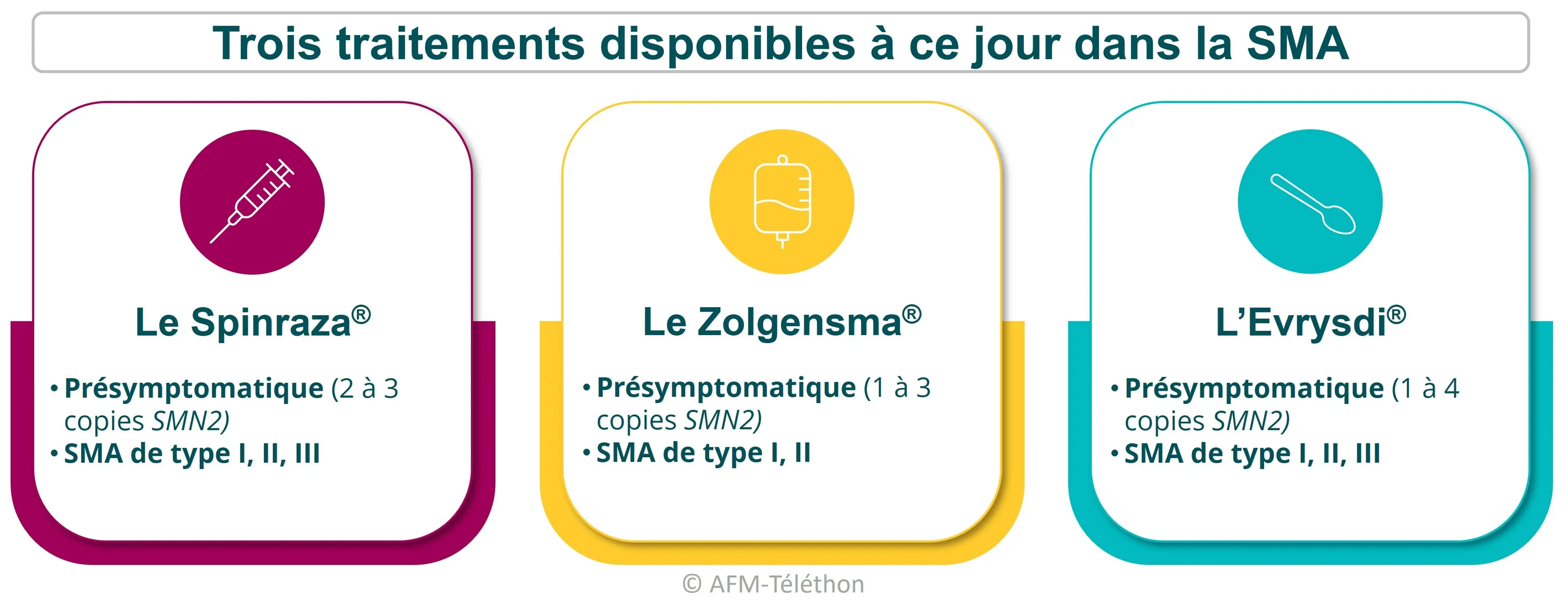
S’ils n’entrainent pas à une guérison complète de la maladie, ils améliorent le plus souvent considérablement la santé des patients et leur qualité de vie. Ils augmentent également leur espérance de vie.
Sans traitement : faiblesse musculaire, qui retentit sur les capacités motrices et la fonction respiratoire.
Avec un traitement après l’apparition des symptômes : effets bénéfiques sur le développement moteur, avec un recul de 6 ans environ (ces données pourront être adaptées sur le long terme).
Avec un traitement avant l’apparition des symptômes (via le dépistage néonatal) : développement normal dans la plupart des cas, avec un recul 4 ans environ (ces données pourront être adaptées sur le long terme).
Comment le type de traitement est-il décidé ?
La mise en place de ces traitements se fait au sein des consultations pluridisciplinaires spécialisées dans les maladies neuromusculaires.
Une RCP pour décider au cas par cas
Une réunion de concertation pluridisciplinaire (RCP) regroupe plusieurs professionnels de santé experts de la maladie afin de choisir la meilleure prise en charge d’un patient en fonction des avancées récentes (publications, données en vie réelle…) et de leur expérience. Dans la SMA, où les traitements sont encore récents, le choix d’un traitement se décide au cas par cas au cours d’une RCP. Ce choix prend notamment en compte l’âge du patient, l’âge du début de la maladie, le type de SMA, le nombre de copies du gène SMN2, les capacités motrices, respiratoires, d’alimentation, la présence ou non d’une arthrodèse…
Lorsque le nourrisson est très symptomatique à la naissance (SMA de type 0), il peut être proposé en RCP et décidé en accord avec les parents de l’enfant que le traitement n’est pas possible.
Le Spinraza®
Commercialisé par le laboratoire Biogen, le Spinraza® (nusinersen) est un oligonulécotide antisens qui cible le gène SMN2 pour lui faire fabriquer la protéine SMN manquante.
Il permet sa réexpression dans les motoneurones avec un bénéfice clinique réel, notamment sur la fonction motrice, d’importance variable selon les personnes, le type de SMA, l’âge de début du traitement…
Des essais cliniques sont toujours en cours pour continuer à évaluer les effets du Spinraza.
Pour en savoir plus, voir la page « Le Spinraza® dans la SMA ».
Le Zolgensma®
Le Zolgensma® (onasemnogene abeparvovec) est un produit de thérapie génique qui vient suppléer le gène SMN1 défectueux par un gène SMN1 thérapeutique afin de fabriquer la protéine SMN, manquante dans la maladie.
Commercialisé par Novartis, il a montré des effets positifs et durables dans le temps : amélioration des performances motrices et respiratoires, allongement de l’espérance de vie… Les bénéfices sont cependant variables d’une personne à l’autre.
Plusieurs essais cliniques continuent d’évaluer ses effets dans différentes formes de SMA (type I, II, stade présymptomatique…). En France, plusieurs études sont en cours : l'essai SPECTRUM, une étude de suivi à long terme du Zolgensma®.
Pour en savoir plus, voir la page « Le Zolgensma® dans la SMA »
L'Evrysdi®
Développé par le laboratoire Roche, l'Evrysdi® (risdiplam) est un modificateur d'épissage qui augmente la quantité de protéine SMN en corrigeant la maturation du gène SMN2. Il améliore la fonction motrice avec une efficacité durable dans le temps, mais variable selon les personnes.
Des essais sont en cours pour continuer d'évaluer les effets de l'Evrysdi®, notamment en présymptomatique chez des nourrissons de moins de 6 semaines (essai RAINBOWFISH) ou de moins de 20 jours (essai PUPFISH).
Pour en savoir plus, voir la page « L’Evrysdi® dans la SMA »
Lire les Avancées dans l’amyotrophie spinale proximale (SMA)
Quelle est la prise en charge en complément des médicaments disponibles ?
L’amyotrophie spinale proximale liée à SMN1 (SMA) dispose désormais d’un arsenal thérapeutique innovant avec le Spinraza®, le Zolgensma® et l’Evrysdi®, qui ciblent la cause génétique de la maladie. Cependant, même s’ils peuvent améliorer considérablement le développement moteur et la qualité de vie des patients, ils ne permettent pas de guérir complètement la maladie.
C’est pourquoi une prise en charge pluridisciplinaire symptomatique, c’est-à-dire ciblant les conséquences de la maladie (symptômes), est nécessaire. Associée à un suivi médical régulier, elle empêche ou retarde la survenue de certaines conséquences de la faiblesse musculaire résiduelle et améliore le confort de vie au quotidien. Elle est adaptée au cas par cas et évolutive, en fonction des atteintes liées à la maladie (musculaire, respiratoire…) et des besoins de chaque personne.
- La prise en charge orthopédique (kinésithérapie...) vise à limiter si nécessaire la fonte musculaire, à préserver la force musculaire et à entretenir la liberté des mouvements.
- Un suivi respiratoire doit être mis en place précocement, dès le diagnostic connu. Lorsqu'il existe une atteinte des muscles respiratoires, celle-ci requiert une prise en charge spécifique.
- En cas de troubles de la parole et/ou de difficultés à avaler, une rééducation spécifique par un orthophoniste limite la gêne qu'ils occasionnent.
- Il est souvent nécessaire d’utiliser des aides techniques pour se déplacer, communiquer, prendre soin de soi…
- Le conseil génétique permet d’évaluer le risque et d'accompagner une personne, ou une famille, confrontée à ce potentiel risque de développer ou de transmettre cette maladie.
La carte d’urgence Amyotrophie spinale infantile, mise en place par la filière FILNEMUS, présente les recommandations importantes pour la prise en charge médicale d’urgence des personnes atteintes d'amyotrophie spinale. Seul un médecin peut la délivrer.
S’informer, pour de meilleurs soins
Le Zoom sur… l’amyotrophie spinale proximale (Octobre 2019) présente une information générale sur la maladie et apporte des réponses concrètes et utiles au quotidien.
Lire le Zoom sur… l’amyotrophie spinale proximale (SMA)
D’autres pistes de recherche
La recherche dans l'amyotrophie spinale infantile (SMA) est intense et féconde depuis plus de vingt ans.
Le saviez-vous ?
La France a joué un rôle essentiel dans la recherche sur la SMA, en particulier avec l'équipe de Judith Melki de l’hôpital Necker (Paris) dont les travaux ont abouti en 1995 à l’identification du gène SMN.
Les chercheurs continuent d’étudier la maladie pour mieux la comprendre : mécanisme moléculaire en cause, rôle de la protéine SMN, marqueurs biologiques…
D’autres médicaments sont en cours de développement, pour agir directement sur la production de protéine SMN, ou sur d’autres cibles (le muscle, la jonction neuromusculaire…) comme des anti-myostatines (apitegromab...)
Lire les Avancées dans l’amyotrophie spinale proximale (SMA)
Un registre français dans l'amyotrophie spinale proximale
Le Registre SMA France a été mis en place début 2020 avec pour objectif de collecter les données de toutes les personnes atteintes d’amyotrophie spinale proximale liée à SMN1 en France.
Aller sur le site internet du registre SMA France
Une étude a mis en évidence l'intérêt de cet outil pour suivre l'évolution de la maladie à l'ère des traitements.
Des congrès scientifiques ou journées d'information sur la maladie
- SMA Europe, qui rassemble plusieurs associations représentatives des personnes atteintes de SMA en Europe, dont l’AFM-Téléthon, organise tous les 2 ans un congrès scientifique international sur l'amyotrophie spinale proximale liée à SMN1. La dernière édition a eu lieu du 12 au 14 mars 2026 à Budapest (Hongrie).
- Un second rendez-vous incontournable est organisé annuellement aux États-Unis par l’association nord-américaine Cure SMA. La dernière édition s'est déroulée du 24 au 26 juin 2026.