Dystrophie musculaire de Duchenne
La myopathie de Duchenne affaiblit progressivement les muscles. En cause, l'absence de dystrophine, une protéine qui leur est essentielle. Elle touche surtout les garçons mais concerne aussi quelques filles. Grâce aux soins et aux médicaments disponibles, près de 10 ans de vie ont été gagnés en vingt ans, mais aujourd'hui, des essais de thérapie génique nourrissent l’espoir de faire bouger encore davantage les lignes !
Des muscles qui s'affaiblissent
La dystrophie musculaire de Duchenne (DMD, myopathie de Duchenne, dystrophie musculaire liée à l’X) provoque une perte de force des muscles, qui survient peu à peu dès l'enfance. Cette maladie rare d'origine génétique touche d’abord les muscles striés squelettiques, ceux des membres, du tronc… et ceux de la respiration comme le diaphragme ou les muscles abdominaux (utiles pour tousser). Elle atteint aussi le cœur (un muscle strié un peu différent) et certains muscles lisses comme ceux de la paroi de l’intestin ou de la vessie.
La dystrophine en cause
Dans la myopathie de Duchenne, des anomalies du gène de la dystrophine, gène DMD situé sur le chromosome X, provoquent l'absence totale de cette protéine. Dans une autre forme de la maladie, la dystrophie musculaire de Becker, la dystrophine est produite en faible quantité ou sous une forme raccourcie, ce qui limite sa sévérité.
La myopathie de Duchenne touche en moyenne 1 sur 5000 naissances de garçons
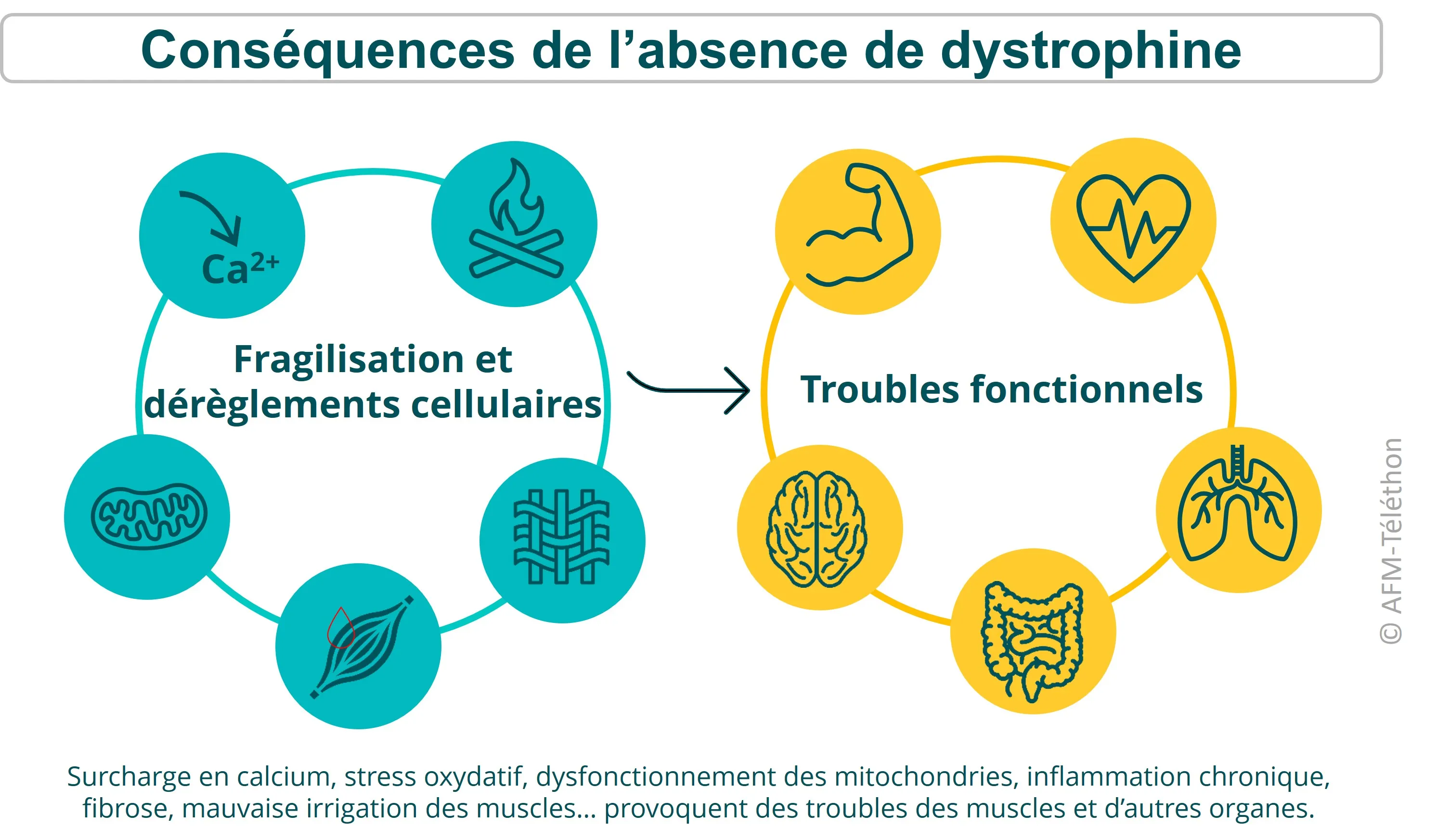
L'absence de dystrophine a de nombreux effets négatifs en cascade sur le fonctionnement des cellules musculaires et les muscles : inflammation, oxydation, perte des mitochondries, fibrose… D’autres fonctions comme la respiration, ou le fonctionnement cardiaque sont progressivement impactées car ils dépendent des muscles. Mais beaucoup d'autres organes et fonctions sont touchés par l'absence de dystrophine.
La transmission de la maladie
La myopathie de Duchenne se manifeste chez les garçons. Les femmes dont le chromosome X porte une anomalie du DMD ne présentent, le plus souvent, pas de gêne même si certaines auront des symptômes légers et quelques autres, une authentique myopathie de Duchenne, similaire à celle des garçons.
Le chromosome X altéré chez une femme peut se transmettre à sa descendance (50 % de risque). Il s’agit d’une transmission dite récessive liée au chromosome X. Les filles qui reçoivent ce chromosome X altéré ne seront pas malades (en général), alors que les garçons oui. Le conseil génétique permet d'informer et d'accompagner une personne, ou une famille, confrontée au risque de développer ou de transmettre cette maladie.
Les signes de la maladie
Les premières difficultés motrices apparaissent dans l'enfance vers l'âge de 3 à 4 ans : les muscles sont moins forts, notamment ceux des jambes, et des difficultés à la marche peuvent déjà exister, ainsi qu'une plus grande fatigabilité.
Au fil des années, apparaît une faiblesse musculaire progressive des jambes puis des bras et du tronc. Se relever, monter les escaliers, puis marcher deviennent difficiles avant de ne plus être possible.
L’atteinte du cœur, un muscle lui aussi, ne se manifeste que bien plus tard, même si elle commence à se développer sans symptômes assez tôt dans l’enfance. Elle aboutit à une cardiomyopathie qui limite l’efficacité du cœur.
Il peut exister aussi chez certains enfants des troubles cognitifs comme un retard ou des troubles du langage, des difficultés d’apprentissage, des troubles de la communication ou comportementaux.
Les médicaments disponibles
Des traitements de fond prescrits tôt dans l'enfance freinent la maladie. Ils protègent :
- les muscles, repoussant de plus de 2 ans en moyenne la perte de la marche ;
- le cœur, retardant l’installation de la cardiomyopathie ;
- la respiration en limitant le développement de l'atteinte respiratoire.
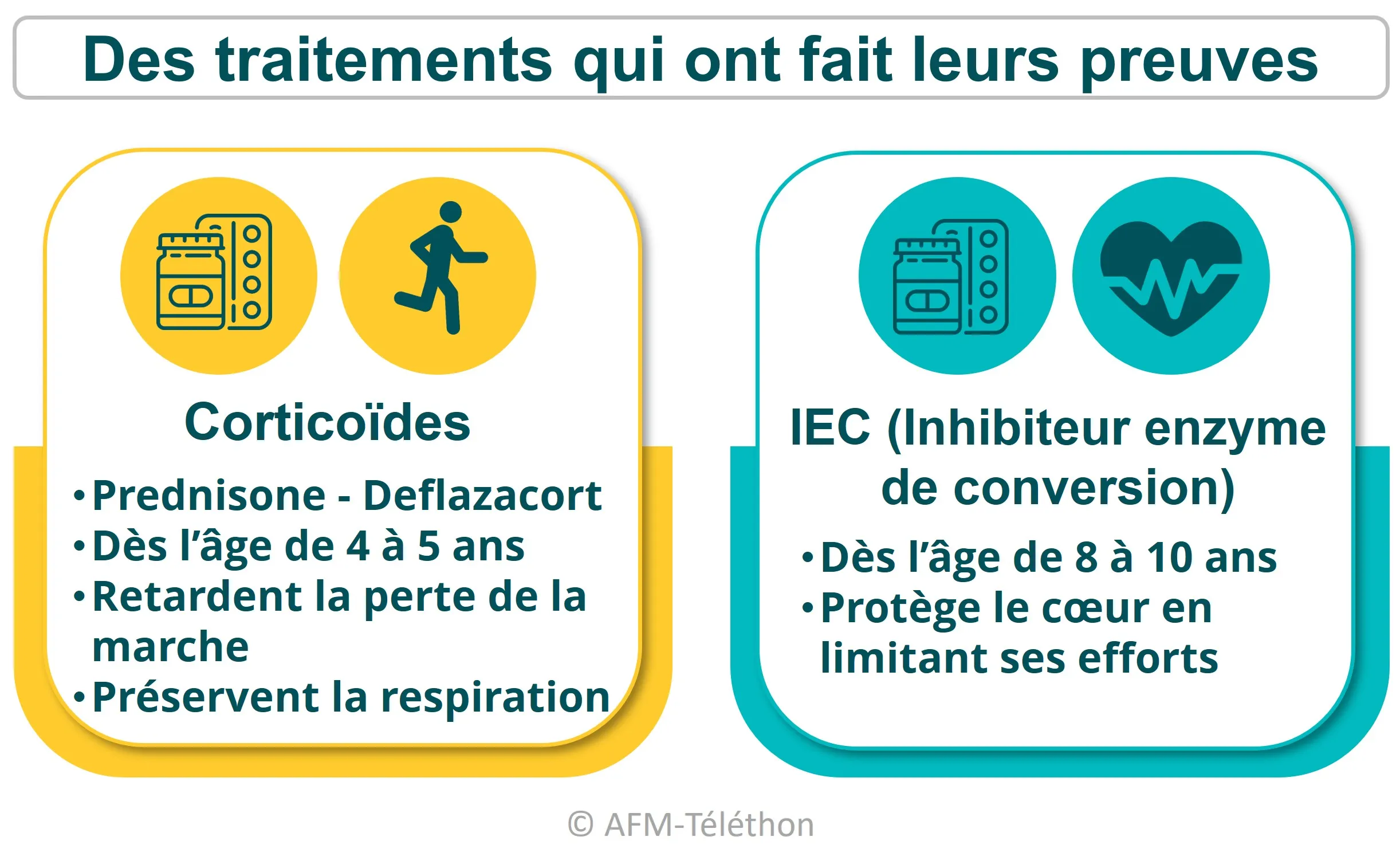
- Les corticoïdes retardent la perte de la marche et préservent les capacités respiratoires. Ils sont prescrits systématiquement entre 4 et 6 ans, une prescription assortie de mesures complémentaires (vitamine D et calcium, suivi diététique...) pour minimiser leurs effets secondaires.
- Les Inhibiteurs de l’enzyme de conversion de l’angiotensine (IEC) sont utilisés systématiquement et précocement (à partir de l'âge de 8 - 10 ans) pour retarder l'apparition des conséquences cardiaques de la DMD. Des bêtabloquants peuvent être proposés en complément devant certains troubles du rythme cardiaque.
Les recommandations en France, sont de prescrire ces traitements assez tôt dans l’enfance, comme le prévoit le Protocole national de diagnostic et de soins (PNDS) dans la dystrophie musculaire de Duchenne, publié en 2019 par la Haute Autorité de Santé (HAS).
Deux nouveaux médicaments dans la DMD
- Le givinostat (Duvyzat) diminue l'inflammation et la fibrose du muscle en agissant sur des molécules qui modulent l'expression de certains gènes (les histones désacétylases (HDAC)). Le Givinostat a reçu une autorisation conditionnelle de mise sur le marché en Europe en juin 2025 chez les patients atteints de DMD, âgés de 6 ans et plus, en capacité de marcher. Mais la Haute autorité de santé (HAS) a rendu le 12 février 2026 un avis défavorable au remboursement du givinostat (Duvyzat®) dans cette indication en France. Cette décision donne un coup d'arrêt à sa commercialisation. Les patients déjà traités pourront continuer de l'être encore quelques mois. Côté essai clinique, le givinostat est aussi en cours d'évaluation chez des patients non-marchants (moins de 18 ans) dans l'essai ULYSSES en France.
- Le vamorolone (Agamree), un corticoïde de synthèse dit "dissociatif" développé récemment, a reçu une autorisation de mise sur le marché en Europe. En France, la Haute autorité de santé (HAS) a estimé qu'il n'apportait pas de bénéfice clinique supplémentaire par rapport aux corticoïdes classiques, tout en étant favorable à son remboursement. Elle a refusé l'autorisation d'accès précoce au médicament. Le médicament n'est donc pas disponible en France.
Fin de l’ataluren (Translarna®) : traitement qui ciblait les anomalies de types codon Stop du gène DMD, l'ataluren n'est plus autorisé à la commercialisation en Europe. Cette décision du 28 mars 2025 de ne pas renouveler l'AMM conditionnelle de l'ataluren repose sur l'absence de preuve d'efficacité sur la marche. Le médicament n'est plus disponible en France : après avis de l'agence de sécurité du médicament (ANSM), les ministres chargés de la santé et de la sécurité sociale ont pris la décision de mettre fin à la prise en charge dérogatoire par les régimes de Sécurité sociale de ce traitement. Ils en ont informé les professionnels de santé susceptible de prescrire ce médicament.
Le registre français des dystrophinopathies
Outils de collecte de données de santé français dédié aux myopathies de Duchenne et de Becker, le Registre francais des dystrophinopathies (registre DYS) est né en 2019. Coordonné par le Pr. Isabelle Desguerre (Hôpital Necker, Paris) et financé par l’AFM-Téléthon, il recense les données cliniques et génétiques des patients atteints de DMD ou de DMB, et les femmes concernées, suivis en consultation en France. Son but est d’alimenter les travaux de recherche et de faciliter l’accès aux études et essais cliniques pour les malades.
Les soins au quotidien
La prise en charge médicale de la maladie comprend des soins réguliers destinés à limiter les complications orthopédiques, cardiaques et respiratoires… Elle repose sur de la rééducation, des traitements pour faciliter la respiration comme la ventilation assistée lorsque cela est nécessaire, la prescription de médicaments (corticoïdes, médicaments pour le cœur) et des interventions chirurgicales si besoin.
Ces soins améliorent le confort et l'espérance de vie des personnes atteintes.
La prise en charge médicale est instaurée dans le cadre d’un suivi médical régulier en consultation pluridisciplinaire neuromusculaire et dès le diagnostic de la maladie. Les soins proposés dépendent de l'évolution de chaque personne. Ils sont réajustés au cours du temps dès que nécessaire.
- Un suivi cardiologique régulier est nécessaire, afin qu'un traitement adapté soit mis en route si des signes cardiaques sont détectés par les examens médicaux en particulier, les IEC dès l’âge de 8-10 ans, puis une prise en charge au cas par cas.
- La prise en charge orthopédique entretient la souplesse et l'amplitude des mouvements ; elle prévient ou limite la progression d'une déformation de la colonne vertébrale (scoliose).
- La prise en charge respiratoire commencée dès les premiers signes d'une atteinte respiratoire permet de conserver des capacités pulmonaires optimales.
- L’utilisation d’aides techniques permet de continuer à réaliser les gestes de la vie quotidienne de manière autonome (fauteuil roulant ou scooter électrique, support de bras…).
L'atteinte cognitive, lorsqu'elle est présente, nécessite une évaluation à l'aide d'un bilan neuropsychologique. Ce dernier permet de repérer les points fort et les difficultés pour proposer une rééducation adaptée et des solutions facilitant notamment la scolarité et la vie sociale.
Faciliter la scolarité. Chaque enfant, quelle que soit sa situation, doit être scolarisé dans les meilleures conditions possibles. Des dispositifs d'accompagnement facilitent la scolarité des enfants en situation de handicap à toutes les étapes de l'apprentissage.
Les indispensables pour scolariser son enfant sur le site de l'AFM-Téléthon
Et en cas d'urgence ? La carte d’urgences Dystrophie musculaire de Duchenne (DMD), mise en place par la filière FILNEMUS donne les recommandations importantes pour la sécurité et la prise en charge médicale d’urgence des personnes atteintes de myopathie de Duchenne.
La recherche d'autres traitements
La recherche de nouveaux traitements très active s'oriente sur trois grandes approches, lesquelles ciblent soit l'origine de la maladie, soit ses conséquences.
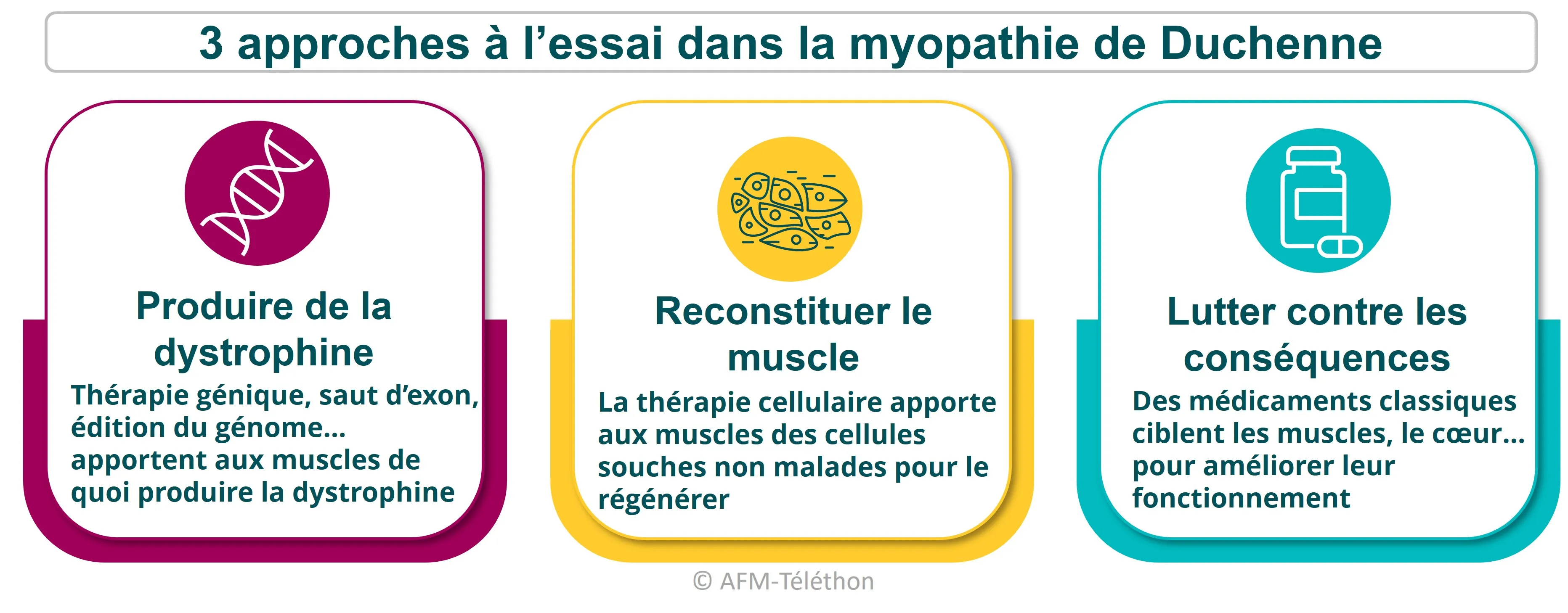
Thérapies innovantes
La thérapie génique, le saut d'exon et la translecture de codon-stop sont des stratégies thérapeutiques innovantes permettant aux muscles de fabriquer des formes de dystrophine fonctionnelles. La thérapie cellulaire est une autre approche qui apporte aux muscles des cellules en bonne santé.
La thérapie génique micro-dystrophine
La thérapie génique consiste à remplacer le gène de la dystrophine (gène DMD) défectueux par un gène thérapeutique codant une microdystrophine, transporté par un vecteur jusqu'aux muscles, le plus souvent un vecteur viral de type AAV. La microdystrophine a été choisie pour ce transfert de gène car le gène DMD entier est trop grand pour le petit virus AAV. Elle contient toutes les parties utiles à sa fonction dans les cellules musculaires.
Cinq produits de thérapie génique en essais cliniques

- Le GNT0004 est évalué dans un essai clinique de phase I/II/III ayant lieu dans des centres français et internationaux. Le recrutement dans la phase pivot de l'essai (phase III) est en cours depuis juillet 2025.
- Le SRP-9001 (Elevidys®) a obtenu aux États-Unis en juin 2023 une autorisation conditionnelle de mise sur le marché, chez les garçons atteints de DMD âgés de 4 à 5 ans, étendue en juin 2024 aux garçons de plus de 4 ans. Le médicament est disponible pour les patients marchants uniquement. Il n'est pas disponible pour les patients non-marchants depuis juillet 2025. En Europe, l'Elevidys™ a reçu un avis défavorable à sa commercialisation chez les garçons âgés de 3 à 7 ans atteints de DMD, de l'Agence européenne du médicament (EMA).
Deux essais du SRP-9001/Elevidys® en France sont suspendus : l'essai ENVOL (son recrutement en France était terminé) et l'essai ENVISION qui était en préparation.
La thérapie par saut d’exon
Le saut d'exon est une technique qui rend possible la fabrication d’une protéine à partir d’un gène défectueux, en agissant sur son ARN messager, au moment de sa formation (maturation). Elle utilise des oligonucléotides antisens, fragments d'ADN ou d’ARN fabriqués en laboratoire, qui en reconnaissant une zone précise de l’ARN messager issu du gène endommagé, permettent de fabriquer à nouveau une protéine.
Plusieurs oligonucléotides antisens ont été conçus (avec différentes structures chimiques) pour cibler les anomalies du gène DMD, comme des suppressions d’un ou plusieurs exons : appelées délétions, elles sont relativement fréquentes (le saut d'exon 51 concerne environ 14 % de patients, le saut d'exon 53 : 10%, le saut d'exon 45 : 9 %).
Quatorze oligonucléotides antisens en essais cliniques dont certains en France :
. SQY51 (saut d'exon 51), un oligonucléotide antisens tricyclo-ADN : essai AVANCE1
. Eteplirsen (saut d'exon 51) : essai MIS51ON
Quatre médicaments de saut d'exon autorisés (AMM conditionnelle) aux États Unis ou Japon.
. Casimersen (Amondys45®/SRP-4045 : saut d'exon 45) (USA)
. Eteplirsen (Exondys51® : saut d'exon 51) (USA)
. Golodirsen (Vyondys53/SRP-4053 : saut d'exon 53) (USA)
. Viltolarsen (Viltepso®/NS-065/NCNP-01 : saut d'exon 53) (Japon et USA).
La thérapie cellulaire
Plusieurs approches sont en développement dans la DMD.
- Le CAP-1002 (Deramiocel) est un produit contenant des cellules souches cardiaques de donneurs, qui après administration à à des patients atteints de DMD favorisent la régénération musculaire cardiaque. Les résultats de l'essai HOPE-2 évaluant le CAP-1002, montrent qu’il est bien toléré et améliore la morphologie et la fibrose cardiaque et ainsi que la fonction des membres supérieurs (développée par le laboratoire Capricor Therapeutics). Suite à la demande de commercialisation du Deramiocel déposée aux Etats-Unis début 2025, l'agence sanitaire américaine, la FDA, s'est prononcée en sa défaveur en juillet 2025 et a demandé des données cliniques additionnelles.
- Une thérapie cellulaire appelée DT-DEC01 repose sur l’utilisation de cellules chimériques exprimant la dystrophine et créées à partir de la fusion de deux myoblastes (les cellules souches précurseurs des cellules musculaires), l’un provenant d’un donneur sain, et donc qui exprime la dystrophine, et l’autre de chaque patient atteint de DMD, qui sera traité ensuite par les cellules chimériques obtenues. Les premiers résultats ont été publiés fin 2023.
Autres candidats médicaments
Encore à l'essai, des médicaments ciblant les conséquences de l'absence de dystrophine dans le muscle sont à l'étude : protéger le cœur, limiter l'atteinte respiratoire, préserver la force musculaire, réduire l'inflammation des muscles, améliorer leur irrigation sanguine, lutter contre l'oxydation des tissus, stimuler l'apport d'énergie de la cellule.
Un nouveau produit prometteur
- L'EDG-5506 (sevasemten) est une petite molécule qui protégerait les fibres musculaires des lésions, en particulier les fibres rapides. Développé par le laboratoire Edgewise Therapeutics, elle est évaluée notamment en France dans la Myopathie de Becker, et dans d'autres essais internationaux dans la DMD (essai LYNX, essai FOX pour les patients déjà traités par thérapie génique).
Ils n'ont pas fait leurs preuves
- Le pamrevlumab (FG-3019) est un anticorps monoclonal dont le développement a été arrêté. Son efficacité n'a pas été démontrée chez les garçons atteints de DMD marchants et non marchants en particulier sur la fibrose et sur la fonction motrice.
- Le tamoxifène a été évalué en France dans l'essai TAMDMD maintenant terminé. Les résultats n'ont pas apporté de preuve de son efficacité dans la DMD, chez les patients marchants et non marchants. Son développement dans cette indication n'est pas poursuivi.
Recherche, conférences et congrès : ça bouge !
Une recherche internationale active
De nombreuses équipes de chercheurs travaillent sur la myopathie de Duchenne, tant pour comprendre les rôles de la dystrophine et les mécanismes biologiques de la maladie que pour trouver de nouveaux médicaments pour la guérir.
Des collaborations nationales et internationales pluridisciplinaires permettent aux chercheurs, généticiens, cliniciens... de capitaliser les connaissances acquises sur la maladie, de les transformer en pistes thérapeutiques et de franchir le cap des essais cliniques. Les associations de malades sont parties prenantes de cet effort de recherche à la fois pour financer des projets de recherche et pour sensibiliser, informer et accompagner les patients autour des essais cliniques.
Voir les Avancées dans les dystrophies musculaires de Duchenne et de Becker.
La recherche en 2025 dans le DMD

Des conférences et des congrès
Ils permettent de partager les progrès scientifiques et médicaux, d'impulser des collaborations, de faire naître des projets de recherche, mais aussi d'aborder d'autres questions pour vivre mieux au quotidien.
- Duchenne Care Conférence - mai 2025 : voir les rediffusions disponibles en accès libre
- Duchenne Day, le 7 septembre 2024 : une journée mondiale annuelle, toujours le 7 septembre, qui aborde des thèmes différents chaque année. A cette occasion, le Groupe d'Intérêt Duchenne/Becker de l'AFM-Téléthon a organisé une journée d'information, disponible en replay sur le site de l'AFM-Téléthon.
- Myology 2024, c'était en avril 2024 à Paris. Organisé par l'AFM-Téléthon, ce fut cette année encore une édition très riche, en particulier dans la myopathie de Duchenne : retrouvez les interviews des chercheurs qui présentent leurs avancées


